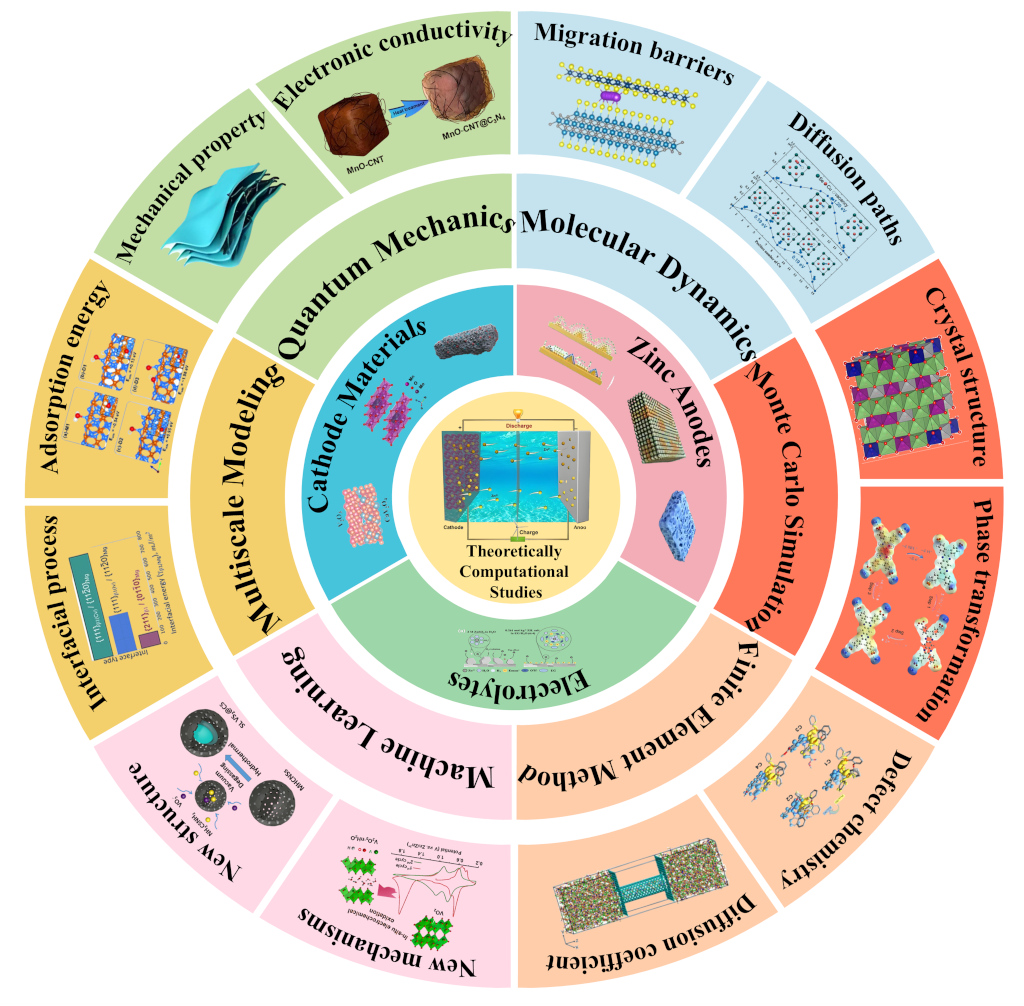
Aqueous zinc-ion secondary batteries (AZIBs) have emerged as a leading candidate for next-generation energy storage due to their high safety, low cost, and environmental compatibility. Despite these advantages, challenges such as zinc dendrite formation, hydrogen evolution reaction (HER), cathode dissolution, and sluggish ion kinetics hinder their widespread commercialization. Theoretical computational studies have become indispensable in addressing these issues by providing atomic-level insights into reaction mechanisms, material properties, and interfacial behaviors. This comprehensive review systematically examines the role of computational methods, including density functional theory (DFT), molecular dynamics (MD), finite element method, machine learning (ML), and multiscale modeling, in advancing AZIBs research. We discuss their applications in cathode material design, anode stabilization, electrolyte optimization, and interfacial engineering. Additionally, we highlight the integration of computational predictions with experimental validations and outline future directions for accelerating AZIBs development.
- Open Access
- Review
Computational Studies for Aqueous Zinc-Ion Secondary Batteries
- Yu Liu 1,
- Yiwen Yuan 1,
- Rui Wang 1,
- Lvhao Duan 1,
- Rongguan Lv 1,
- Yingna Chang 1,
- Huayu Wu 1,
- Xiaoli Yan 1,
- Rong Xing 1, *,
- Rudolf Holze 2, 3, 4, *
Author Information
Received: 15 Sep 2025 | Revised: 22 Oct 2025 | Accepted: 28 Nov 2025 | Published: 09 Dec 2025
Abstract
Graphical Abstract
Keywords
aqueous zinc ion battery | computational studies | density functional theory | molecular dynamic | machine learning
References
- 1.
Zhou, W.; Tang, Y.; Zhang, X.; et al. MOF derived metal oxide composites and their applications in energy storage. Coordin. Chem. Rev. 2023, 477, 214949.
- 2.
Kang, Z.; Zhao, Y.; Yang, D. Review of oil shale in-situ conversion technology. Appl. Energy 2020, 269, 115121.
- 3.
Liu, Y.; Qian, J.; Shi, Y.; et al. Latest advances of metal-organic frameworks-based materials for supercapacitors. Sustain. Mater. Techno. 2023, 36, e00588.
- 4.
Yang, S.; Cheng, Y.; Xiao, X.; et al. Development and application of carbon fiber in batteries. Chem. Eng. J. 2020, 384, 123294.
- 5.
Xiao, X.; Zou, L.; Pang, H.; et al. Synthesis of micro/nanoscaled metal-organic frameworks and their direct electrochemical applications. Chem. Soc. Rev. 2020, 49, 301.
- 6.
Ma, W.; Zhang, Y.; Pan, S.; et al. Smart fibers for energy conversion and storage. Chem. Soc. Rev. 2021, 50, 7009.
- 7.
Yang, Y.; Hoang, M.T.; Bhardwaj, A.; et al. Perovskite solar cells based self-charging power packs: Fundamentals, applications and challenges. Nano Energy 2022, 94, 106910.
- 8.
Yu, F.; Wang, Y.; Liu, Y.; et al. An aqueous rechargeable zinc-ion battery on basis of an organic pigment. Rare Met. 2022, 41, 2230.
- 9.
Peng, Y.; Xu, J.; Xu, J.M.; et al. Metal-organic framework (MOF) composites as promising materials for energy storage applications. Adv. Colloid Interf. 2022, 307, 102732.
- 10.
Xu, Y.; Li, Q.; Pang, H. Recent advances in metal organic frameworks and their composites for batteries. Nano Futures 2020, 4, 032007.
- 11.
Luo, Y.; Tang, Y.; Zheng, S.; et al. Dual anode materials for lithium- and sodium-ion batteries. J. Mater. Chem. A 2018, 6, 4236.
- 12.
Chi, C.; Liu, Z.; Lu, X.; et al. Balance of sulfur doping content and conductivity of hard carbon anode for high-performance K-ion storage. Energy Storage Mater. 2023, 54, 668.
- 13.
Wang, Q.; Liu, Y.; Chen, P. Phenazine-based organic cathode for aqueous zinc secondary batteries. J. Power Sources 2020, 468, 228401.
- 14.
Lv, T.; Peng, Y.; Zhang, G.; et al. How about vanadium-based compounds as cathode materials for aqueous zinc ion batteries. Adv. Sci. 2023, 10, 2206907.
- 15.
Zhang, X.; Zhang, H.; Chen, M.; et al. Zinc-based fiber-shaped rechargeable batteries: Insights into structures, electrodes, and electrolytes. Nano Res. 2025, 18, 94907025.
- 16.
Liu, Y.; Zhi, J.; Hoang, T.K.A.; et al. Paraffin based cathode-electrolyte interface for highly reversible aqueous zinc-ion battery. ACS Appl. Energy Mater. 2022, 5, 4840.
- 17.
Wang, Q.; Liu, Y.; Wang, C.; et al. Vat orange 7 as an organic electrode with ultrafast hydronium-ion storage and super-long life for rechargeable aqueous zinc batteries. Chem. Eng. J. 2023, 451, 138776.
- 18.
Hariprakash, B.; Martha, S.; Ambalavanan, S.; et al. Comparative study of lead-acid batteries for photovoltaic stand-alone lighting systems. J. Appl. Electrochem. 2008, 38, 77.
- 19.
Liu, Y.; Wen, Z.; Wu, X.; et al. Holze. An acid-free rechargeable battery based on PbSO4 and spinel LiMn2O4. Chem. Commun. 2014, 50, 13714.
- 20.
Wang, H.; Liang, Y.; Gong, M.; et al. An ultrafast nickel-iron battery from strongly coupled inorganic nanoparticle/nanocarbon hybrid materials. Nat. Commun. 2012, 3, 917.
- 21.
Liu, Y.; Xie, D.; Shi, Y.; et al. An aqueous rechargeable Fe//LiMn2O4 hybrid battery with superior electrochemical performance beyond mainstream Fe-based batteries. Nano Res. 2024, 17, 5168.
- 22.
Sun, Y.; Liu, Y.; Han, Y.; et al. Effects of thermal transformation on graphene-like lamellar porous carbon and surface-contributed capacitance. Mater. Today Commun. 2021, 29, 102982.
- 23.
Liu, Y.; Xu, Y.; Chang, Y.; et al. A new high-current electrochemical capacitor using MnO2-coated vapor-grown carbon fibers. Crystals 2022, 12, 1444.
- 24.
Baig, M.M.; Khan, S.A.; Ahmad, H.; et al. 3D printing of hydrogels for flexible micro-supercapacitors. FlexMat 2024, 1, 79.
- 25.
Zhu, G.; Hou, Y.; Lu, J.; et al. MXene decorated 3D-printed carbon black-based electrodes for solid-state micro-supercapacitors. J. Mater. Chem. A 2023, 11, 25422.
- 26.
Liu, Y.; Zhang, B.; Wang, F.; et al. Nanostructured intercalation compounds as cathode materials for supercapacitors. Pure Appl. Chem. 2014, 86, 593.
- 27.
Zhang, Y.; Li, Q.; Feng, W.; et al. Valence engineering via polyoxometalate-induced on vanadium centers for efficient aqueous zinc-ion batteries. Angew. Chem. Int. Ed. 2025, 137, e202501728.
- 28.
Liu, Y.; Zhi, J.; Sedighi, M.; et al. Mn2+ ions confined by electrode microskin for aqueous battery beyond intercalation capacity. Adv. Energy Mater. 2020, 10, 2002578.
- 29.
Hu, Q.; Hu, J.; Ma, F.; et al. Redistributing zinc-ion flux by work function chemistry toward stabilized and durable Zn metal batteries. Energy Environ. Sci. 2024, 17, 2554.
- 30.
Liu, Y.; Shi, Q.; Wu, Y.; et al. Highly efficient dendrite suppressor and corrosion inhibitor based on gelatin/Mn2+ co-additives for aqueous rechargeable zinc-manganese dioxide battery. Chem. Eng. J. 2021, 407, 127189.
- 31.
Zhang, Y.; Li, X.; Fan, L.; et al. Ultrathin and super-rough membrane for anti-dendrite separator in aqueous zinc-ion batteries. Cell Rep. Phys. Sci. 2022, 3, 100824.
- 32.
Yang, X.; Wu, W.; Liu, Y.; et al. Chitosan modified filter paper separators with specific ion adsorption to inhibit side reactions and induce uniform Zn deposition for aqueous Zn batteries. Chem. Eng. J. 2022, 450, 137902.
- 33.
Li, H.; Yang, G.; Chen, J.; et al. Revealing the electrochemistry in a voltaic cell by in situ electron microscopy. ChemElectroChem 2022, 9, e202200441.
- 34.
Dong, X.; Wang, Y.; Xia, Y. Re-building daniell cell with a Li-ion exchange film. Sci. Rep. 2014, 4, 6916.
- 35.
Wruck, W.J.; Reichman, B.; Bullock, K.R.; et al. Rechargeable Zn‐MnO2 alkaline batteries. J. Electrochem. Soc. 1991, 138, 3560.
- 36.
Pan, J.; Xu, Y.Y.; Yang, H.; et al. Advanced architectures and relatives of air electrodes in Zn–air batteries. Adv. Sci. 2018, 5, 1700691.
- 37.
Han, M.; Chen, D.; Lu, Q.; et al. Aqueous rechargeable Zn–iodine batteries: Issues, strategies and perspectives. Small 2024, 20, 2310293.
- 38.
Mahmood, A.; Zheng, Z.; Zinc, Y.C. Zinc–bromine batteries: Challenges, prospective solutions, and future. Adv. Sci. 2024, 11, 2305561.
- 39.
Lu, H.; Hu, J.; Zhang, K.; et al. Microfluidic-assisted 3D printing zinc powder anode with 2D conductive MOF/MXene heterostructures for high-stable zinc−organic battery. Adv. Mater. 2024, 36, 2309753.
- 40.
Lu, J.; Wang, T.; Yang, J.; et al. Multifunctional self-assembled bio-interfacial layers for high-performance zinc metal anodes. Angew. Chem. Int. Ed. 2024, 63, e202409838.
- 41.
Ding, X.; Jia, X.; Yuan, H.; et al. Tumor microenvironment–activated Zn//MnO2 battery for sustained and local electrochemical immunotherapy. Sci. Adv. 2025, 11, eadu1647.
- 42.
Brunk, E.; Ursula, R. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem. Rev. 2015, 115, 6217.
- 43.
Song, L.; De, N.; Yan, C.; et al. Correlating solid electrolyte interphase composition with dendrite-free and long life-span lithium metal batteries via advanced characterizations and simulations. Small Methods 2023, 7, 2300168.
- 44.
Tian, M.; Wang, Z.; Yang, H.Y.; et al. Recent progress in computational materials science boosting development of rechargeable batteries. Adv. Energy Mater. 2025, 15, 2403443.
- 45.
Yan, L.; Su, J.; Sun, C.; et al. Review of the first principles calculations and the design of cathode materials for Li-ion batteries. Adv. Manuf. 2014, 2, 358.
- 46.
Markov, I.L. Limits on fundamental limits to computation. Nature 2014, 512, 147.
- 47.
Tuckerman, M.E.; Martyna, G.J. Understanding modern molecular dynamics: Techniques and applications. J. Phys. Chem. B 2000, 104, 159.
- 48.
Yamakov, V.; Wolf, D.; Phillpot, S.R.; et al. Deformation-mechanism map for nanocrystalline metals by molecular-dynamics simulation. Nat. Mater. 2004, 3, 43.
- 49.
van Gunsteren, W.F.; Oostenbrink, C. Methods for classical-mechanical molecular simulation in chemistry: Achievements, limitations, perspectives. J. Chem. Inf. Model. 2024, 64, 6281.
- 50.
Gavilán-Arriazu, E.M.; Mercer, M.P.; Barraco, D.E.; et al. Kinetic monte carlo simulations applied to Li-ion and post Li-ion batteries: A key link in the multi-scale chain. Prog. Energy 2021, 3, 042001.
- 51.
Wu, S.; Chen, Y.; Luan, W.; et al. A review of multiscale mechanical failures in lithium-ion batteries: Implications for performance, lifetime and safety. Electrochem. Energy Rev. 2024, 7, 35.
- 52.
Zhang, J. Modern monte carlo methods for efficient uncertainty quantification and propagation: A survey. Wires. Comput. Stat. 2021, 13, e1539.
- 53.
Zuo, S.; Yin, B.; Xu, Y.; et al. A simplified method of soft connected battery module for finite element method model of battery pack. Int. J. Energy Res. 2021, 45, 10546.
- 54.
Song, S.; Zhu, M.; Xiong, Y.; et al. Mechanical failure mechanism of silicon-based composite anodes under overdischarging conditions based on finite element analysis. ACS Appl. Mater. Interfaces 2021, 13, 34157.
- 55.
Liu, Y.; Zheng, G.; Letov, N.; et al. A survey of modeling and optimization methods for multi-scale heterogeneous lattice structures. J. Mechan. Des. 2021, 143, 040803.
- 56.
Kotsiantis, S.B.; Zaharakis, I.D.; Pintelas, P.E. Machine learning: A review of classification and combining techniques. Artif. Intell. Rev. 2006, 26, 159.
- 57.
Nozarijouybari, Z.; Fathy, H.K. Machine learning for battery systems applications: Progress, challenges, and opportunities. J. Power Sources 2024, 601, 234272.
- 58.
Nair, M.R.; Roy, T. Role of artificial intelligence in the design and discovery of next-generation battery electrolytes. Chem. Phys. Rev. 2025, 6, 011311.
- 59.
Roman, D.; Saxena, S.; Robu, V.; et al. Machine learning pipeline for battery state-of-health estimation. Nat. Mach. Intell. 2021, 3, 447.
- 60.
Parmar, J.; Chouhan, S.; Raychoudhury, V.; et al. Open-world machine learning: Applications, challenges, and opportunities. ACM Comput. Surv. 2023, 55, 1–37.
- 61.
Liu, X.; Zhang, L.; Yu, H.; et al. Bridging multiscale characterization technologies and digital modeling to evaluate lithium battery full lifecycle. Adv. Energy Mater. 2022, 12, 2200889.
- 62.
Chen, X.; Liu, X.; Shen, X.; et al. Applying machine learning to rechargeable batteries: From the microscale to the macroscale. Angew. Chem. Int. Ed. 2021, 133, 24558.
- 63.
de Carvalho, F.O.; Sousa, O.; Assali, L.V.C.; et al. Accelerating Cathode design for zinc-ion batteries using data-driven screening and ab initio calculations. J. Mater. Chem. A 2025, 13, 29317–29322. https://doi.org/10.1039/D5TA02667A.
- 64.
Li, G.; Sun, L.; Zhang, S; et al. Developing cathode materials for aqueous zinc ion batteries: Challenges and practical prospects. Adv. Funct. Mater. 2024, 34, 2301291.
- 65.
Cao, H.; Zheng, Z.; Norby, P.; et al. Electrochemically induced phase transition in V3O7·H2O nanobelts/reduced graphene oxide composites for aqueous zinc-ion batteries. Small 2021, 17, 2100558.
- 66.
Chen, S.; Kong, Y.; Tang, C.; et al. Doping regulation stabilizing δ-MnO2 cathode for high-performance aqueous aluminum-ion batteries. Small 2024, 20, 2312229.
- 67.
Liang, J.; Zhao, Y.; Ren, L.; et al. Dual anions doping enhanced conductivity and stability of layered δ-MnO2 cathode for aqueous zinc-ion battery. Adv. Funct. Mater. 2025, 35, 2501135.
- 68.
Wang, S.; Yao, S.; Dai, N.; et al. Spin symmetry breaking-induced hubbard gap near-closure in N-coordinated MnO2 for enhanced aqueous zinc-ion battery performance. Angew. Chem. Int. Ed. 2024, 63, e202408414.
- 69.
Chao, D.; Ye, C.; Xie, F.; et al. Atomic engineering catalyzed MnO2 electrolysis kinetics for a hybrid aqueous battery with high power and energy density. Adv. Mater. 2020, 32, 2001894.
- 70.
Nam, K.W.; Kim, H.; Choi, J.H.; et al. Crystal water for high performance layered manganese oxide cathodes in aqueous rechargeable zinc batteries. Energy Environ. Sci. 2019, 12, 1999.
- 71.
Chen, H.; Ruan, P.; Zhang, H.; et al. Achieving highly reversible Mn2+/MnO2 conversion reaction in electrolytic Zn-MnO2 batteries via electrochemical-chemical process regulation. Angew. Chem. Int. Ed. 2025, 64, e202423999.
- 72.
Chen, N.; Wang, W.; Ma, Y.; et al. Aqueous zinc-chlorine battery modulated by a MnO2 redox adsorbent. Small Methods 2023, 8, 2201553.
- 73.
Chen, C.; Shi, M.; Zhao, Y.; et al. Al-intercalated MnO2 cathode with reversible phase transition for aqueous Zn-ion batteries. Chem. Eng. J. 2021, 422, 130375.
- 74.
Li, X.; Zhou, Q.; Yang, Z.; et al. Unraveling the role of nitrogen-doped carbon nanowires incorporated with MnO2 nanosheets as high performance cathode for zinc-ion batteries. Energy Environ. Mater. 2022, 6, e12378.
- 75.
Zheng, W.; Cui, Z.; Liu, C.; et al. Tailoring hierarchical MnO2 nanostructures on self-supporting cathodes for high-mass-loading zinc-ion batteries. Chem. Sci. 2024, 15, 20303.
- 76.
Li, M.; Liu, C.; Meng, J.; et al. Hydroxylated manganese oxide cathode for stable aqueous zinc-ion batteries. Adv. Funct. Mater. 2024, 34, 2405659.
- 77.
Xu, N.; Zhang, Y.; Wang, M.; et al. High-performing rechargeable/flexible zinc-air batteries by coordinated hierarchical Bi-metallic electrocatalyst and heterostructure anion exchange membrane. Nano Energy 2019, 65, 104021.
- 78.
Le, T.; Takeuchi, E.S.; Takeuchi, K.J.; et al. Tuning discharge behavior of hollandite α-MnO2 in hydrated zinc ion battery by transition metal substitution. J. Phys. Chem. C 2023, 127, 907.
- 79.
Jiang, H.; Gong, W.; Zhang, Y.; et al. Quench-tailored Al-doped V2O5 nanomaterials for efficient aqueous zinc-ion batteries. J. Energy Chem. 2022, 70, 52.
- 80.
Liu, Y.; Lu, C.; Yang, Y.; et al. Multiple cations nanoconfinement in ultrathin V2O5 nanosheets enables ultrafast ion diffusion kinetics toward high-performance zinc ion battery. Adv. Mater. 2024, 36, 2312982.
- 81.
Wang, W.; Zhang, L.; Duan, Z.; et al. Joint cationic and anionic redox chemistry in a vanadium oxide cathode for zinc batteries achieving high energy density. Carbon Energy 2024, 6, e577.
- 82.
Xu, X.; Qian, Y.; Wang, C.; et al. Enhanced charge transfer and reaction kinetics of vanadium pentoxide for zinc storage via nitrogen interstitial doping. Chem. Eng. J. 2023, 451, 138770.
- 83.
Zhang, B.; Han, X.; Kang, W.; et al. Structure and oxygen-defect regulation of hydrated vanadium oxide for enhanced zinc ion storage via interlayer doping strategy. Nano Res. 2023, 16, 6094.
- 84.
Zhang, Y.; Li, Z.; Liu, M.; et al. Construction of novel polyaniline-intercalated hierarchical porous V2O5 nanobelts with enhanced diffusion kinetics and ultra-stable cyclability for aqueous zinc-ion batteries. Chem. Eng. J. 2023, 463, 142425.
- 85.
Pan, R.; Zheng, A.; He, B.; et al. In situ crafting of a 3D N-doped carbon/defect-rich V2O5-xnH2O nanosheet composite for high performance fibrous flexible Zn-ion batteries. Nanoscale Horiz. 2022, 7, 1501.
- 86.
Wu, T.; Zhu, K.; Qin, C.; et al. Unraveling the role of structural water in bilayer V2O5 during Zn2+-intercalation: Insights from DFT calculations. J. Mater. Chem. A 2019, 7, 5612.
- 87.
Wang, Z.; Liang, P.; Zhang, R.; et al. Oxygen-defective V2O5 nanosheets boosting 3D diffusion and reversible storage of zinc ion for aqueous zinc-ion batteries. Appl. Surf. Sci. 2021, 562, 150196.
- 88.
Chen, J.; Zhang, W.; Zhang, X.; et al. Sodium pre-intercalated carbon/V2O5 constructed by sustainable sodium lignosulfonate for stable cathodes in zinc ion batteries: A comprehensive study. ChemSusChem 2022, 15, e202200732.
- 89.
Wang, Y.; Wei, S.; Qi, Z.; et al. Intercalant-induced V t2g orbital occupation in vanadium oxide cathode toward fast-charging aqueous zinc-ion batteries. Proc. Natl. Acad. Sci. USA 2023, 120, e2217208120.
- 90.
Deng, W.; Li, C.; Zou, W.; et al. Understanding the super-theoretical capacity behavior of VO2 in aqueous Zn batteries. Small 2023, 20, 2309527.
- 91.
Wang, Z.; Cui, P.; Wang, X.; et al. Co-substitution engineering boosting the kinetics and stability of VO2 for Zn ion batteries. Adv. Funct. Mater. 2024, 34, 2407925.
- 92.
Li, Z.; Ren, Y.; Mo, L.; et al. Impacts of oxygen vacancies on zinc ion intercalation in VO2. ACS Nano 2020, 14, 5581.
- 93.
Luo, H.; Wang, B.; Wang, C.; et al. Synergistic deficiency and heterojunction engineering boosted VO2 redox kinetics for aqueous zinc-ion batteries with superior comprehensive performance. Energy Storage Mater. 2020, 33, 390.
- 94.
Luo, P.; Zhang, W.; Cai, W.; et al. Accelerated ion/electron transport kinetics and increased active sites via local internal electric fields in heterostructured VO2-carbon cloth for enhanced zinc-ion storage. Nano Res. 2023, 16, 503.
- 95.
Zhang, W.; Liu, J.; Cai, W.; et al. An. Engineering d-p orbital hybridization through regulation of interband energy separation for durable aqueous Zn//VO2(B) batteries. Chem. Eng. J. 2023, 464, 142711.
- 96.
Guan, K.; Duan, K.; Yang, G.; et al. Ultra-long cycle H-doped VO2(B) cathode for high capacity aqueous Zn-ion battery. Mater. Today Adv. 2022, 14, 100230.
- 97.
Liu, W.; Zong, H.; Li, M.; et al. Ta4C3 modulated MOF-derived 3D crosslinking network of VO2(B)@Ta4C3 for high-performance aqueous zinc ion batteries. ACS Appl. Mater. Interfaces 2023, 15, 13554.
- 98.
Zhang, L.; Fang, D.; Wang, F.; et al. nterlayer and O-vacancy engineering co-boosting fast kinetics and stable structure of hydrated sodium ammonium vanadate for aqueous zinc-ion battery. Chem. Eng. J. 2025, 506, 159920.
- 99.
Cui, F.; Wang, D.; Hu, F.; et al. Deficiency and surface engineering boosting electronic and ionic kinetics in NH4V4O10 for high-performance aqueous zinc-ion battery. Energy Storage Mater. 2022, 44, 197.
- 100.
Li, M.; Zhang, Y.; Hu, J.; et al. Universal multifunctional hydrogen bond network construction strategy for enhanced aqueous Zn2+/proton hybrid batteries. Nano Energy 2022, 100, 107539.
- 101.
Liu, Y.; Lv, J.; Cao, T.; et al. Unsaturated Ni/V centers and short Ni·V/Ni distances in nickel vanadate for high-performance zinc-ion battery. Chem. Eng. J. 2022, 441, 136007.
- 102.
Wang, S.; Cao, Y.; Qin, J.; et al. Revealing the self-generated heterointerface of NaV2O5 in Zn storage via a scalable production method. ACS Sustain. Chem. Eng. 2023, 11, 6710.
- 103.
Zang, Q.; Cheng, X.; Chen, S.; et al. Oxygen defect engineering triggered by S-doping boosts the performance of H2V3O8 nanobelts for aqueous Zn-ion storage. Chem. Eng. J. 2023, 452, 139396.
- 104.
Cao, H.; Peng, C.; Zheng, Z.; et al. Orientation effect of zinc vanadate cathode on zinc ion storage performance. Electrochim. Acta 2021, 388, 138646.
- 105.
Liu, C.; Liu, Y.; Liu, X.; et al. Coordination polymer-derived Al3+-doped V2O3/C with rich oxygen vacancies for an advanced aqueous zinc-ion battery with ultrahigh rate capability. Sustain. Energy Fuels 2022, 6, 2020.
- 106.
Bai, Y.; Zhang, H.; Xiang, B.; et al. Oxygen vacancy-rich, binder-free copper pyrovanadate for zinc ion storage. Chem. Eng. J. 2021, 420, 130474.
- 107.
Li, Q.; Zhang, Q.; Zhou, Z.; et al. Boosting Zn-ion storage capability of self-standing Zn-doped Co3O4 nanowire array as advanced cathodes for high-performance wearable aqueous rechargeable Co//Zn batteries. Nano Res. 2021, 14, 91.
- 108.
Long, J.; Gu, J.; Yang, Z.; et al. Highly porous, low band-gap NixMn3xO4 (0.55 < x < 1.2) spinel nanoparticles with in situ coated carbon as advanced cathode materials for zinc-ion batteries. J. Mater. Chem. A 2019, 7, 17854.
- 109.
Mallick, S.; Choutipalli, V.S.K.; Bag, S.; et al. Defect engineered ternary spinel: An efficient cathode for an aqueous rechargeable zinc-ion battery of long-term cyclability. ACS Appl. Mater. Interfaces 2022, 14, 37577.
- 110.
Shao, T.; Zhang, Y.; Cao, T.; et al. Structural regulation of ZnMn2O4 cathode material by K, Fe-double doping to improve its rate and cycling stability for rechargeable aqueous zinc-based batteries. Chem. Eng. J. 2022, 431, 133735.
- 111.
Tan, Y.; He, J.; Wang, B.; et al. Tuning the layer structure of molybdenum trioxide towards high-performance aqueous zinc-ion batteries. Chin. Chem. Lett. 2023, 34, 107410.
- 112.
Teng, C.; Yang, F.; Sun, M.; et al. Structural and defect engineering of cobaltosic oxide nanoarchitectures as an ultrahigh energy density and super durable cathode for Zn-based batteries. Chem. Sci. 2019, 10, 7600.
- 113.
Wang, K.; Guo, G.; Tan, X.; et al. Achieving high-energy and long-cycling aqueous zinc-metal batteries by highly reversible insertion mechanisms in Ti-substituted Na0.44MnO2 cathode. Chem. Eng. J. 2023, 451, 139059.
- 114.
Wei, J.; Guo, J.; Wang, S.; et al. Fabrication of dual-functional electrodes using oxygen vacancy abundant NiCo2O4 nanosheets for advanced hybrid supercapacitors and Zn-ion batteries. Inorg. Chem. Front. 2022, 9, 4452.
- 115.
Zhang, Y.; Xu, J.; Liu, C.; et al. Ion-exchange-induced high-performance of inverse spinel Mg2VO4 for aqueous zinc-ion batteries. J. Power Sources 2022, 549, 232075.
- 116.
Bai, Y.; Luo, L.; Song, W.; et al. Nitrogen-vacancy-rich VN clusters embedded in carbon matrix for high-performance zinc ion batteries. Adv. Sci. 2024, 11, 2308668.
- 117.
Bian, Y.; Hu, R.; Wang, H.; et al. Developing an in situ polyoxovanadate to vanadium nitride/carbon conversion strategies in manufacture aqueous zinc ion batteries cathode with ultrahigh rate capability. Adv. Funct. Mater. 2024, 35, 2418671.
- 118.
Chen, J.; Guo, K.; Ren, T.; et al. Aluminium-doped vanadium nitride as cathode material for high-performance aqueous zinc-ion batteries. J. Power Sources 2025, 626, 235751.
- 119.
Dai, Y.; Lu, R.; Zhang, C.; et al. Zn2+-mediated catalysis for fast-charging aqueous Zn-ion batteries. Nat. Catal. 2024, 7, 776.
- 120.
Luo, S.; Cui, J.; Liang, S.; et al. Carbon-enveloped pea-shaped vanadium nitride nanorods for aqueous zinc ion batteries. J. Mater. Chem. C 2024, 12, 6153.
- 121.
Posadzy, M.; Garcia-Rodriguez, M.; Flores-Lasluisa, J.X.; et al. High-performance transition metal nitride/carbon nanofiber composites as positive electrode in rechargeable Zn-air batteries. Carbon 2025, 239, 120308.
- 122.
Yao, X.; Khanam, Z.; Li, C.; et al. Unlatching the additional zinc storage ability of vanadium nitride nanocrystallites. Small 2024, 20, 2312036.
- 123.
Wang, H.; Hou, W.; Wang, X.; et al. Construction of MOF-derived vanadium nitride decorated micro/mesoporous carbon nanofibers for aqueous zinc-ion batteries. Chem. Eur. J. 2025, 31, e202403903.
- 124.
Chen, D.; Lu, M.; Wang, B.; et al. Uncover the mystery of high-performance aqueous zinc-ion batteries constructed by oxygen-doped vanadium nitride cathode: Cationic conversion reaction works. Energy Storage Mater. 2021, 35, 679.
- 125.
Jung, H.; Lee, J.; Park, J.; et al. A mesoporous tungsten oxynitride nanoffbers/graphite felt composite electrode with high catalytic activity for the cathode in Zn-Br flow battery. Small 2023, 19, 2208280.
- 126.
Dong, C.; Tang, S.; Chen, Y.; et al. Identifying the synergistic Na+/Zn2+ cointercalation mechanism for boosting electrochemical performance of Na4Fe3(PO4)2P2O7 in Zn-ion batteries. ACS Mater. Lett. 2023, 5, 1170.
- 127.
Hu, L.; Wu, Z.; Lu, C.; et al. Principles of interlayer-spacing regulation of layered vanadium phosphates for superior zinc-ion batteries. Energy Environ. Sci. 2021, 14, 4095.
- 128.
Wu, Z.; Lu, C.; Ye, F.; et al. Bilayered VOPO4·2H2O nanosheets with high-concentration oxygen vacancies for high-performance aqueous zinc-ion batteries. Adv. Funct. Mater. 2021, 31, 2106816.
- 129.
Zhao, S.; Qu, G.; Wang, C.; et al. Towards advanced aqueous zinc battery by exploiting synergistic effects between crystalline phosphide and amorphous phosphate. Nanoscale 2021, 13, 18586.
- 130.
Zhao, D.; Chen, S.; Lai, Y.; et al. A stable “rocking-chair” zinc-ion battery boosted by low-strain Zn3V4(PO4)6 cathode. Nano Energy 2022, 100, 107520.
- 131.
Chen, L.; Gao, X.; Nie, H.; et al. Electrochemical oxidation endows VN/V3S4 heterostructure with high performance in aqueous zinc-ion batteries. J. Alloy. Compd. 2023, 958, 170491.
- 132.
Li, X.; Zhao, S.; Qu, G.; et al. Defect engineering in Co-doped Ni3S2 nanosheets as cathode for high-performance aqueous zinc ion battery. J. Mater. Sci. Techn. 2022, 118, 190.
- 133.
Tian, G.; Ling, D.; Chen, Z.; et al. Effective CuO/Cu7S4 nanospheres heterostructures for advanced “rocking-chair” zinc-ion battery. J. Colloid Interf. Sci. 2025, 679, 334.
- 134.
Li, C.; Liu, C.; Wang, Y.; et al. Drastically-enlarged interlayer-spacing MoS2 nanocages by inserted carbon motifs as high performance cathodes for aqueous zinc-ion batteries. Energy Storage Mater. 2022, 49, 144.
- 135.
Liu, F.; Li, L.; Xu, S.; et al. Cobalt-doped MoS2·nH2O nanosheets induced heterogeneous phases as high-rate capability and long-term cyclability cathodes for wearable zinc-ion batteries. Energy Storage Mater. 2023, 55, 1–11.
- 136.
Li, S.; Huang, C.; Gao, L.; et al. Unveiling the “proton lubricant” chemistry in aqueous zinc-MoS2 batteries. Angew. Chem. Int. Ed. 2022, 134, e202211478.
- 137.
Liu, L.; Yang, W.; Chen, H.; et al. High zinc-ion intercalation reaction activity of MoS2 cathode based on regulation of thermodynamic metastability and interlayer water. Electrochim. Acta 2022, 410, 140016.
- 138.
Liu, J.; Xu, P.; Liang, J.; et al. Boosting aqueous zinc-ion storage in MoS2 via controllable phase. Chem. Eng. J. 2020, 389, 124405.
- 139.
Mao, Y.; Bai, J.; Si, J.; et al. Magneto-electrochemistry driven ultralong-life Zn-VS2 aqueous zinc-ion batteries. Mater. Horiz. 2023, 10, 3162.
- 140.
Ren, Z.; Sun, Y.; Yin, Y.; et al. Metallic V5S8 microparticles with tunnel-like structure for high-rate and stable zinc-ion energy storage. Energy Storage Mater. 2021, 42, 786.
- 141.
Tan, Y.; Li, S.; Zhao, X.; et al. Unexpected role of the interlayer “dead Zn2+” in strengthening the nanostructures of VS2 cathodes for high-performance aqueous Zn-ion storage. Adv. Energy Mater. 2022, 12, 2104001.
- 142.
Wang, X.; Zhang, S.; Yang, R.; et al. Hierarchical carbon nanosheet confined defective MoSx cathode towards long-cycling zinc-ion-battery. Nano Res. 2023, 16, 9364.
- 143.
Xu, H.; Yang, W.; Liu, H.; et al. Boosting kinetics of tellurium redox reaction for high-performance aqueous zinc-tellurium batteries. Chem. Eng. J. 2023, 465, 142896.
- 144.
Yu, J.; Cai, D.; Si, J.; et al. MOF-derived NiCo2S4 and carbon hybrid hollow spheres compactly concatenated by electrospun carbon nanofibers as self-standing electrodes for aqueous alkaline Zn batteries. J. Mater. Chem. A 2022, 10, 4100.
- 145.
Bai, Y.; Zhang, H.; Song, H.; et al. Engineering anion defects of ternary V-S-Se layered cathodes for ultrafast zinc ion storage. Nano Energy 2024, 120, 109090.
- 146.
Chen, X.; Zhang, A.; Zou, H.; et al. Defect engineering modulated MoSe2 cathode achieves highly effective photo-responsive zinc ion battery. Energy Storage Mater. 2024, 70, 103457.
- 147.
Aghdam, A.M.; Habibzadeh, S.; Javanbakht, M.; et al. High interspace-layer manganese selenide nanorods as a high performance cathode for aqueous zinc-ion batteries. ACS Appl. Energy Mater. 2023, 6, 3225.
- 148.
Wu, Z.; Lu, C.; Wang, Y.; et al. Ultrathin VSe2 nanosheets with fast ion diffusion and robust structural stability for rechargeable zinc-ion battery cathode. Small 2020, 16, 2000698.
- 149.
Cui, M.; Zhu, Y.; Lei, H.; et al. Anion–cation competition chemistry for comprehensive high performance prussian blue analogs cathodes. Angew. Chem. Int. Ed. 2024, 63, e202405428.
- 150.
Fu, H.; Wang, X.; Ye, L.; et al. Optimizing Fe in Mn-based prussian blue analogs with dual redox-active sites to enhance operating voltage and durability in Zn-ion batteries. Chem. Eng. J. 2025, 506, 160308.
- 151.
Li, W.; Hei, P.; Sai, Y.; et al. Vanadium-based prussian blue analogue for high energy aqueous zinc-iodine batteries. Chem. Eng. J. 2025, 510, 161720.
- 152.
Puthiyaveetil, P.P.; Nair, A.; Dilwale, S.; et al. Insights on prussian blue analogue cathode material engineered with polypyrrole surface protection layer for aqueous rechargeable zinc metal battery. Small 2024, 21, 2409947.
- 153.
Qian, Y.; Chang, G.; Huang, C.; et al. A transformed prussian blue analog as host of I2 for long-life aqueous zinc-iodine battery. Chem. Eng. J. 2025, 503, 158392.
- 154.
Wang, L.; Liu, N.; Li, Q.; et al. Dual redox reactions of silver hexacyanoferrate prussian blue analogue enable superior electrochemical performance for zinc-ion storage. Angew. Chem. Int. Ed. 2025, 137, e202416392.
- 155.
Gao, W.; Cheng, S.; Zhang, Y.; et al. Efficient charge storage in zinc–iodine batteries based on pre-embedded iodine-ions with reduced electrochemical reaction barrier and suppression of polyiodide self-shuttle effect. Adv. Funct. Mater. 2023, 33, 2211979.
- 156.
Tan, Y.; Yang, H.; Miao, C.; et al. Hydroxylation strategy unlocking multi-redox reaction of manganese hexacyanoferrate for aqueous zinc-ion battery. Chem. Eng. J. 2023, 457, 141323.
- 157.
Ding, C.; Zhao, Y.; Yin, W.; et al. Regulating intermolecular hydrogen bonds in organic cathode materials to realize ultra-stable, flexible and low-temperature aqueous zinc-organic batteries. Angew. Chem. Int. Ed. 2025, 137, e202417988.
- 158.
Hong, H.; Wang, Y.; Wei, Z.; et al. Constructing a janus catholyte/cathode structure: A new strategy for stable Zn-organic batteries. Adv. Mater. 2024, 36, 2410209.
- 159.
Hua, K.; Ma, Q.; Liu, Y.; et al. High-performance bipolar small-molecule organic cathode for wide-temperature range aqueous zinc-ion batteries. ACS Nano 2025, 19, 14249.
- 160.
Li, C.; Hu, L.; Ren, X.; et al. Asymmetric charge distribution of active centers in small molecule quinone cathode boosts high-energy and high-rate aqueous Zinc-organic batteries. Adv. Funct. Mater. 2023, 34, 2313241.
- 161.
Zhang, L.; Wang, X.; Wang, X.; et al. Electron-withdrawing group functionalization for improved zinc-ion storage in organic electrode materials. Chem. Eng. J. 2025, 521, 166985.
- 162.
Zhang, Y.; Li, M.; Li, Z.; et al. A high capacity p-type organic cathode material for aqueous zinc batteries. Angew. Chem. 2024, 136, e202410342.
- 163.
Kundu, D.; Oberholzer, P.; Glaros, C.; et al. An organic cathode for aqueous Zn batteries: Taming a unique phase evolution towards stable electrochemical cycling. Chem. Mater. 2018, 30, 3874.
- 164.
Li, L.; Wang, Y.; Gong, W.; et al. Building stable small molecule imide cathodes toward ultralong-life aqueous zinc-organic batteries. Chem. Eng. J. 2023, 465, 142824.
- 165.
Wang, Y.; Wang, X.; Tang, J.; et al. A quinoxalinophenazinedione covalent triazine framework for boosted high-performance aqueous zinc-ion batteries. J. Mater. Chem. A 2022, 10, 13868.
- 166.
Wang, J.; Zhang, X.; Yan, Z.; et al. Tetrathiafulvalene as a sustainable cathode with high rate and long life-span for aqueous Zinc-ion battery at low temperatures. Chem. Eng. J. 2023, 459, 141649.
- 167.
Ding, Y.; Cai, C.; Ma, L.; et al. Tailoring MnO2 cathode interface via organic–inorganic hybridization engineering for ultra-stable aqueous zinc-ion batteries. Adv. Energy Mater. 2024, 15, 2402819.
- 168.
Sang, B.; Wang, X.; Feng, K.; et al. Boosting zinc-ion storage performance by interlayer chemistry modulation on an organic-inorganic hybrid cathode. J. Colloid Interf. Sci. 2024, 653, 199.
- 169.
Luo, Z.; Liu, Z.; He, H.; et al. Suppressing the dissolution of vanadium by organic-inorganic hybrid for aqueous zinc-ion batteries. J. Mater. Sci. Techn. 2023, 145, 93.
- 170.
Ma, X.; Cao, X.; Yao, M.; et al. Organic–inorganic hybrid cathode with dual energy storage mechanism for ultrahigh-rate and ultralong-life aqueous zinc-ion batteries. Adv. Mater. 2022, 34, 2105452.
- 171.
Nagarai, R.; Pakhira, S.; Aruchamy, K.; et al. Catalyzing the intercalation storage capacity of aqueous zinc-ion battery constructed with Zn(II) preinserted organo-vanadyl hybrid cathode. ACS Appl. Energy Mater. 2020, 3, 3425.
- 172.
Sariyer, S.; Yesilot, S.; Kilic, N.; et al. Polyphosphazene based inorganic-organic hybrid cathode containing pyrene tetraone sides for aqueous zinc-ion batteries. Batter. Supercaps 2023, 6, e202200529.
- 173.
Cao, J.; Wu, H.; Zhang, D.; et al. In-situ ultrafast construction of zinc tungstate interface layer for highly reversible zinc anodes. Angew. Chem. Int. Ed. 2024, 63, e202319661.
- 174.
Gao, G.; Huo, X.; Li, B.; et al. Customizing water-scarce, zinc ion-rich Helmholtz plane of zinc anode for Ah-scale Zn metal batteries. Energy Environ. Sci. 2024, 17, 7850.
- 175.
Huang, W.; Huang, Y.; Huang, X.; et al. 3D leaf-like copper-zinc alloy enables dendrite-free zinc anode for ultra-long life aqueous zinc batteries. Small 2024, 20, 2404294.
- 176.
Ling, W.; Nie, C.; Wu, X.; et al. Ion sieve interface assisted zinc anode with high zinc utilization and ultralong cycle life for 61 Wh/kg mild aqueous pouch battery. ACS Nano 2024, 18, 5003.
- 177.
Peng, Z.; Yan, H.; Zhang, Q.; et al. Stabilizing zinc anode through ion selection sieving for aqueous Zn-ion batteries. Nano Lett. 2024, 24, 9137.
- 178.
Shu, C.; An, Y.; Liu, Y.; et al. Construction of corrosion-resistant and dendrite-free zinc anode by coating nano-ceriumoxide for highly stable zinc battery. Chem. Eng. J. 2025, 509, 161096.
- 179.
Zhao, M.; Lv, Y.; Qi, J.; et al. Crystallographic reorientation induced by gradient solid-electrolyte interphase for highly stable zinc anode. Adv. Mater. 2024, 36, 2412667.
- 180.
Cao, Z.; Zhang, H.; Song, B.; et al. Angstrom-level ionic sieve 2D-MOF membrane for high power aqueous zinc anode. Adv. Funct. Mater. 2023, 33, 2300339.
- 181.
Hu, Z.; Zhou, L.; Meng, D.; et al. Surface engineering for ultrathin metal anodes enabling high performance Zn-ion batteries. ACS Appl. Mater. Interfaces 2023, 15, 5161.
- 182.
Lee, J.; Kim, R.; Kim, S.; et al. Dendrite-free Zn electrodeposition triggered by interatomic orbital hybridization of Zn and single vacancy carbon defects for aqueous Zn-based flow batteries. Energy Environ. Sci. 2020, 13, 2839.
- 183.
Luo, Y.; Hu, J.; Cai, S.; et al. Chelate-capped nano-AgZn3 dual interphase remodeling the local environment for reversible dendrite-free zinc anode. Small 2023, 19, 2303268.
- 184.
Pan, Z.; Cao, Q.; Gong, W.; et al. Zincophilic 3D ZnOHF nanowire arrays with ordered and continuous Zn2+ ion modulation layer enable long-term stable Zn metal anodes. Energy Storage Mater. 2022, 50, 435.
- 185.
Qin, H.; Kuang, W.; Huang, D.; et al. Achieving high-rate and high-capacity Zn metal anodes via a three-in-one carbon protective layer. J. Mater. Chem. A 2022, 10, 17440.
- 186.
Liu, P.; Zhang, Z.; Hao, R.; et al. Ultra-highly stable zinc metal anode via 3D-printed g-C3N4 modulating interface for long life energy storage systems. Chem. Eng. J. 2021, 403, 126425.
- 187.
Song, Q.; Liang, J.; Liu, S.; et al. Negatively charged insulated boron nitride nanofibers directing subsurface zinc deposition for dendrite-free zinc anodes. Nano Res. 2023, 16, 403.
- 188.
Chen, Z.; Shen, T.; Xiao, X.; et al. An ultrahigh-modulus hydrogel electrolyte for dendrite-free zinc ion batteries. Adv. Mater. 2024, 36, 2413268.
- 189.
Guo, S.; Yan, M.; Xu, D.; et al. Anti-freezing hydrogel electrolyte with regulated hydrogen bond network enables high-rate and long cycling zinc batteries. Energy Environ. Sci. 2025, 18, 418.
- 190.
Li, J.; Zhang, H.; Liu, Z.; et al. Boosting dendrite-free zinc anode with strongly polar functional group terminated hydrogel electrolyte for high-safe aqueous zinc-ion batteries. Adv. Funct. Mater. 2024, 35, 2412865.
- 191.
Luo, F.; Yang, S.; Wu, Q.; et al. Hydrogel electrolytes with an electron/ion dual regulation mechanism for highly reversible flexible zinc batteries. Energy Environ. Sci. 2024, 17, 8570.
- 192.
Shen, Z.; Liu, Y.; Li, Z.; et al. Highly-entangled hydrogel electrolyte for fast charging/discharging properties in aqueous zinc ion batteries. Adv. Funct. Mater. 2024, 35, 2406620.
- 193.
Shen, Z.; Zhai, Z.; Liu, Y.; et al. Hydrogel electrolytes based rechargeable zinc ion batteries under harsh conditions. Nano-Micro Lett. 2025, 17, 227.
- 194.
Wang, Y.; Liang, B.; Li, D.; et al. Hydrogel electrolyte design for long-lifespan aqueous zinc batteries to realize a 99% Coulombic efficiency at 90 °C. Joule 2025, 9, 101944.
- 195.
Xiang, Z.; Li, Y.; Cheng, X.; et al. Lean-water hydrogel electrolyte with improved ion conductivity for dendrite-free zinc-Ion batteries. Chem. Eng. J. 2024, 490, 151524.
- 196.
Xia, H.; Zhang, W.; Miao, C.; et al. Ultra-thin amphiphilic hydrogel electrolyte for flexible zinc-ion paper batteries. Energy Environ. Sci. 2024, 17, 6507.
- 197.
Xiong, Y.; Cheng, H.; Jiang, Y.; et al. A novel water-reducer-based hydrogel electrolyte for robust and flexible Zn-I2 battery. Energy Storage Mater. 2025, 74, 103981.
- 198.
Xu, X.; Li, S.; Yang, S.; et al. Superelastic hydrogel electrolyte incorporating helical protein molecules as zinc ion transport pathways to enhance cycling stability of zinc metal batteries. Energy Environ. Sci. 2024, 17, 7919.
- 199.
Abbasi, A.; Xu, Y.; Abouzari-Lotf, E.; et al. Phosphonated graphene oxide-modified polyacrylamide hydrogel electrolytes for solid-state zinc-ion batteries. Electrochim. Acta 2022, 435, 141365.
- 200.
Chen, Y.; Zhang, Z.; Cai, P.; et al. Polyoxotungstate featuring zinc-ion-triggered structural transformation as an efficient electrolyte additive for aqueous zinc-ion batteries. Angew. Chem. Int. Ed. 2024, 137, e202420284.
- 201.
Dong, J.; Zhou, G.; Ding, W.; et al. Machine learning-assisted benign transformation of three zinc states in zinc ion batteries. Energy Environ. Sci. 2025, 18, 4872.
- 202.
Feng, W.; Zhang, L.; Cheng, Y.; et al. Screening and design of aqueous zinc battery electrolytes based on the multimodal optimization of molecular simulation. J. Phys. Chem. Lett. 2025, 16, 3326.
- 203.
Huang, S.; Fu, H.; Kwon, H.M.; et al. Stereoisomerism of multi-functional electrolyte additives for initially anodeless aqueous zinc metal batteries. Nat. Commun. 2025, 16, 6117.
- 204.
Luo, H.; Gou, Q.; Zheng, Y.; et al. Machine learning-assisted high-donor number electrolyte additive screening toward construction of dendrite-free aqueous zinc-ion batteries. ACS Nano 2025, 19, 2427.
- 205.
Shang, Y.; Kundi, V.; Pal, I.; et al. Highly potent and low-volume concentration additives for durable aqueous zinc batteries: Machine learning-enabled performance rationalization. Adv. Mater. 2024, 36, 2309212.
- 206.
Wu, Q.; Huang, J.; Zhang, J.; et al. Multifunctional cellulose nanocrystals electrolyte additive enable ultrahigh-rate and dendrite-free Zn anodes for rechargeable aqueous zinc batteries. Angew. Chem. Int. Ed. 2024, 63, e202319051.
- 207.
Xie, Z.; Chen, N.; Zhang, M.; et al. Carbonate-assisted chaotropic electrolyte for zinc ion battery with wide temperature operation. ACS Energy Lett. 2024, 9, 3380.
- 208.
Chen, Y.; Gong, F.; Deng, W.; et al. Dual-function electrolyte additive enabling simultaneous electrode interface and coordination environment regulation for zinc-ion batteries. Energy Storage Mater. 2023, 58, 20.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com