Myocardial lipotoxicity is a fundamental pathological mechanism responsible for myocardial injury associated with various metabolic disorders such as obesity and type 2 diabetes. This condition arises from an imbalance in lipid metabolic homeostasis, leading to the excessive accumulation of toxic lipid intermediates in cardiomyocytes, which ultimately results in structural and functional impairments of the myocardium. This review provides a comprehensive overview of the mechanisms by which myocardial lipotoxicity occurs and its significant involvement in cardiovascular diseases, including heart failure, arrhythmias, and atherosclerosis. The myocardium primarily acquires exogenous fatty acids through albumin-bound free fatty acids and lipoprotein lipase-mediated lipolysis, facilitated by transporter proteins like CD36 and FATP. When the uptake of fatty acids surpasses oxidative capacity, toxic lipids such as ceramides and DAG accumulate, disrupting essential signaling pathways—including AMPK, PPAR, PKC, and NF-κB. This disruption triggers mitochondrial dysfunction, endoplasmic reticulum stress, oxidative stress, inflammatory responses, and various forms of cell death. Together, these mechanisms lead to myocardial remodeling, abnormalities in electrical activity, and the onset and progression of vascular pathologies. The article also reviews current therapeutic strategies aimed at mitigating myocardial lipotoxicity, including lifestyle modifications and pharmacological interventions such as SGLT2 inhibitors and trimetazidine. Furthermore, it explores future research directions, focusing on ceramide synthesis, inflammatory pathways, and personalized medicine, with the goal of providing a theoretical foundation and innovative insights for the clinical prevention and treatment of lipotoxicity-related cardiovascular diseases.
- Open Access
- Review
Myocardial Lipotoxicity in Cardiovascular Diseases: Mechanisms, Therapeutic Strategies, and Drug Target Discovery
- Yaxin Xu 1,2,
- Xinrui Li 3,
- Lu Fu 1,2,*
Author Information
Received: 09 Oct 2025 | Revised: 11 Nov 2025 | Accepted: 13 Nov 2025 | Published: 15 May 2026
Abstract
Keywords
myocardial lipotoxicity | fatty acid metabolism | heart failure | arrhythmia | atherosclerosis | therapeutic strategies
1. Introduction
The heart, as a high-energy-demand organ, relies on a continuous and efficient supply of adenosine triphosphate (ATP) to sustain its contractile function. Under normal physiological conditions, the adult mammalian heart exhibits remarkable metabolic flexibility, deriving approximately 60–70% of its ATP from the β-oxidation of fatty acids, with the remainder supplied primarily by glucose and lactate oxidation [1,2]. This intricate balance of substrate utilization is tightly regulated to meet dynamic energy demands. However, in the context of pervasive metabolic disorders such as obesity and type 2 diabetes, this homeostatic balance is profoundly disrupted. A hallmark of these conditions is elevated circulating levels of free fatty acids, which drive excessive lipid uptake into cardiomyocytes. When the influx of fatty acids surpasses the oxidative and storage capacities of the heart, it leads to the pathological accumulation of toxic lipid intermediates, including ceramides and diacylglycerols (DAG)—a condition termed myocardial lipotoxicity [3,4,5].
Myocardial lipotoxicity is now recognized as a fundamental pathological mechanism contributing to a broad spectrum of cardiovascular diseases (CVDs). It initiates a deleterious cascade of cellular events, including mitochondrial dysfunction, endoplasmic reticulum (ER) stress, oxidative stress, chronic inflammation, and various forms of programmed cell death [6,7]. These processes collectively promote myocardial structural remodeling, electrical instability, and vascular dysfunction, thereby driving the pathogenesis and progression of heart failure, arrhythmias, and atherosclerosis [8,9,10]. Despite significant advances in understanding its core mechanisms, the intricate molecular networks and signaling pathways involved in lipotoxicity are not fully elucidated, and specific therapeutic strategies targeting this process remain an area of intense investigation.
This review aims to provide a comprehensive and updated overview of the mechanisms underlying myocardial lipotoxicity and its central role in cardiovascular pathology. We will first delineate the sources and transport mechanisms of myocardial fatty acids, setting the stage for understanding how lipid overload occurs. Subsequently, we will delve into the pathophysiological mechanisms, detailing the generation of toxic lipid species, organelle dysfunction, and the ensuing inflammatory and cell death responses. The impact of lipotoxicity on specific cardiovascular diseases—heart failure, arrhythmias, and atherosclerosis—will be systematically examined. Finally, we will summarize current and emerging therapeutic strategies and propose future research directions, with the goal of offering a foundational framework for developing novel interventions to mitigate the burden of lipotoxicity-related cardiovascular diseases.
2. Sources and Transport Mechanisms of Myocardial Fatty Acids
2.1. Sources of Myocardial Fatty Acids
Cardiomyocytes primarily obtain fatty acids from the circulatory system, which can be categorized into exogenous and endogenous sources, with exogenous sources being the predominant contributors [1]. Exogenous fatty acids are those absorbed by the myocardium from the blood, mainly through two pathways: First, the direct uptake of free fatty acids (FFAs) bound to albumin [11]. Due to their hydrophobicity nature, most long-chain fatty acids in the blood associate with plasma albumin, resulting in only a small fraction remaining free. This binding not only enhances the solubility and transport capacity of fatty acids but also mitigates their potential cytotoxic effects. Cardiomyocytes can efficiently uptake fatty acids directly from albumin-FFA complexes, making this the primary source [12]. Second, fatty acids are released through the enzymatic hydrolysis of circulating lipoproteins. The myocardium can access fatty acids by breaking down triglyceride (TG)-rich lipoproteins, predominantly chylomicrons from dietary fats and very-low-density lipoprotein (VLDL) produced by the liver [13,14]. This process is facilitated by two key lipases: Lipoprotein lipase (LPL), the primary enzyme in this process, is anchored to the endothelial surface of myocardial capillaries. As chylomicrons and VLDL traverse the cardiac vasculature, LPL efficiently hydrolyzes their TG, releasing FFAs and glycerol [15]. These newly generated FFAs, being in close proximity to cardiomyocytes, are rapidly taken up and utilized. The activity and expression of LPL are crucial regulatory points for myocardial fatty acid supply; its deficiency is closely linked to hypertriglyceridemia, myocardial dysfunction, and even heart failure [16,17,18]. The second key enzyme, endothelial lipase (EL), which belongs to the same family as LPL and is synthesized by vascular endothelial cells [19,20], primarily impacts high-density lipoprotein (HDL) metabolism. However, studies have shown that under certain pathological conditions (e.g., early heart failure when LPL activity may decline), EL expression significantly increases, potentially serving as an alternative source for myocardial fatty acid acquisition to compensate for energy supply deficits [21]. Additionally, cardiomyocytes can store fatty acids, with excess fatty acids being esterified into TG and stored in cytoplasmic lipid droplets. When energy demand increases or exogenous supply is insufficient, these stored TG can be hydrolyzed by intracellular lipases, releasing fatty acids directly into metabolic pathways to provide an emergency energy reserve [22].
2.2. Transport of Myocardial Fatty Acids
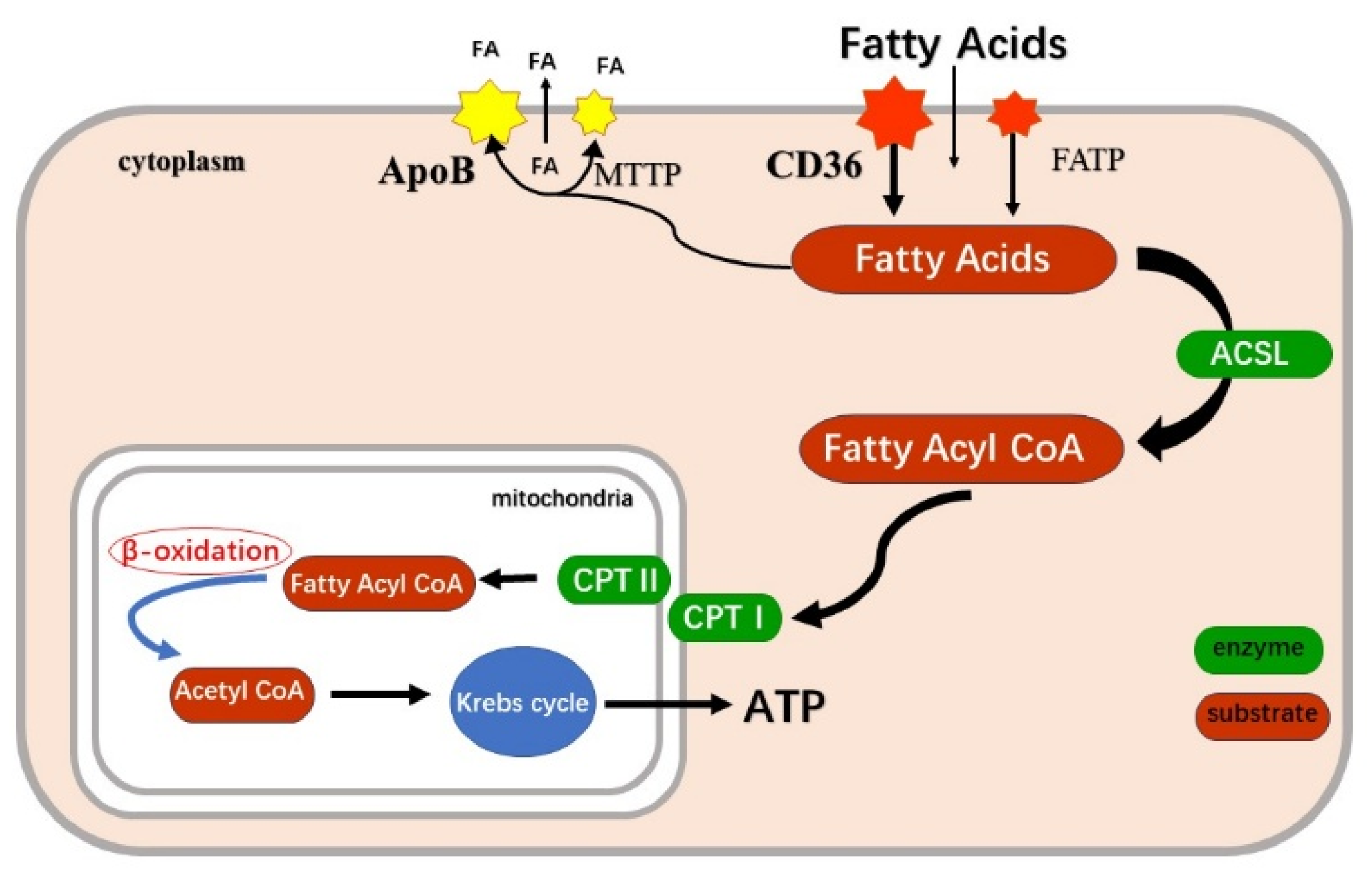
The transport of fatty acids from the bloodstream into cardiomyocytes requires crossing the hydrophobic barrier of the cell membrane. This process is not merely passive diffusion; rather, it is a highly regulated, saturable, and efficient mechanism mediated by various transporter proteins [2]. The early “flip-flop” model proposed that protonated fatty acids first integrate into the outer leaflet of the cell membrane, flip to the inner leaflet, and then dissociate inside the cell. However, the role of passive diffusion is relatively minor, especially under physiological concentrations or during periods of high energy demand when fatty acid flux is substantial; passive diffusion alone cannot meet myocardial requirements [23,24]. The currently accepted primary mode of transport is protein-mediated. This mechanism relies on specific carrier proteins embedded within the cell membrane, characterized by their saturability, high affinity, and specificity [25,26,27]. CD36 (also known as Fatty Acid Translocase, FAT) is the most functionally significant and well-studied fatty acid transporter in the myocardium, accounting for 50–60% of total myocardial fatty acid uptake [14]. As a transmembrane glycoprotein, it not only facilitates fatty acid transport but also engages in signal transduction, with its activity dynamically regulated. In response to signals such as insulin stimulation or muscle contraction, CD36 stored in intracellular vesicles rapidly translocates to the plasma membrane, significantly enhancing the myocardial capacity for fatty acid uptake within a short period [28]. The Fatty Acid Transport Protein family FATPs, comprising at least six members (FATP1-6), includes FATP1 and FATP6 which also contribute to fatty acid transport in the myocardium [11]. These proteins not only facilitate the translocation of fatty acids across the membrane but some members also exhibit acyl-CoA synthetase activity, potentially coupling transport and activation. Additionally, the membrane-associated Fatty Acid Binding Protein (FABPpm) has been identified as playing a role in fatty acid absorption in cardiomyocytes [29]. Together, these three proteins form a critical network for fatty acid transport across the cardiomyocyte membrane, ensuring that the heart efficiently and flexibly acquires the energy substrates needed under varying physiological states. Once inside the cytoplasm, fatty acids cannot diffuse freely; they require a series of “chaperoning” and “gating” mechanisms to be accurately delivered to their final destination—the mitochondrial matrix for β-oxidation.
FFAs entering the cytosol pose potential “lipotoxicity” risks and may disrupt cellular function. Heart-type Fatty Acid Binding Protein (H-FABP or FABP3) plays a crucial role in addressing this challenge [30]. As a highly abundant small soluble protein in the cardiomyocyte cytoplasm, H-FABP’s core functions include binding and solubilization, targeted transport, as well as buffering and protection [31]. Long-chain fatty acids cannot directly cross the inner mitochondrial membrane; their entry into the mitochondrial matrix relies on a complex enzymatic transport system known as the Carnitine Shuttle System. Carnitine Palmitoyltransferase (CPT) is the system’s core component, and this process is also the rate-limiting step for long-chain fatty acid oxidation [32]. This system operates through the coordinated efforts of three key components: Initially, fatty acids are activated on the outer mitochondrial membrane by Long-Chain Acyl-CoA Synthetase (ACSL), producing high-energy acyl-CoA. Carnitine Palmitoyltransferase I (CPT-I), located on the outer mitochondrial membrane, serves as the key regulatory enzyme, catalyzing the transfer of the acyl group from acyl-CoA to carnitine, forming acylcarnitine. This acylcarnitine is then translocated across the inner mitochondrial membrane into the matrix via Carnitine-Acylcarnitine Translocase (CACT). Inside the matrix, Carnitine Palmitoyltransferase II (CPT-II), situated on the matrix side of the inner mitochondrial membrane, transfers the acyl group back from carnitine to CoA, thereby regenerating acyl-CoA. At this stage, the fatty acid has successfully entered the mitochondrial matrix and can immediately enter the β-oxidation cycle [33,34,35].
In summary, the sourcing and transport of myocardial fatty acids is a complex biological process that encompasses macroscopic circulation and microscopic molecular interactions, involving multiple steps and stringent regulation. It originates from fatty acids carried by albumin or lipoproteins in the bloodstream, which are processed by enzymes such as LPL and EL, and subsequently taken up by cardiomyocytes via the coordinated action of membrane proteins like CD36 and FATP [12,16]. Within cardiomyocytes, H-FABP regulates safe and efficient “escorting” fatty acids to the mitochondria, where the critical “gatekeeping” function of the carnitine shuttle system ultimately delivers them into the “workshop” of β-oxidation [35]. This intricate process is finely regulated at various levels, including substrate concentration, hormonal signals, and gene transcription, ensuring cardiac energy homeostasis under diverse physiological conditions. However, in pathological states such as diabetes, obesity, and heart failure, this regulatory network is disrupted, leading to an imbalance in fatty acid metabolism that can initiate or exacerbate myocardial injury [36]. Consequently, conducting in-depth research into the molecular mechanisms and regulatory networks governing myocardial fatty acid transport and metabolism not only enhances our understanding of cardiac energy metabolism but also provides a vital theoretical foundation and potential therapeutic targets for developing novel strategies to combat cardiovascular diseases associated with metabolic disorders (Figure 1).
3. Pathophysiological Mechanisms and Regulatory Targets of Myocardial Lipotoxicity
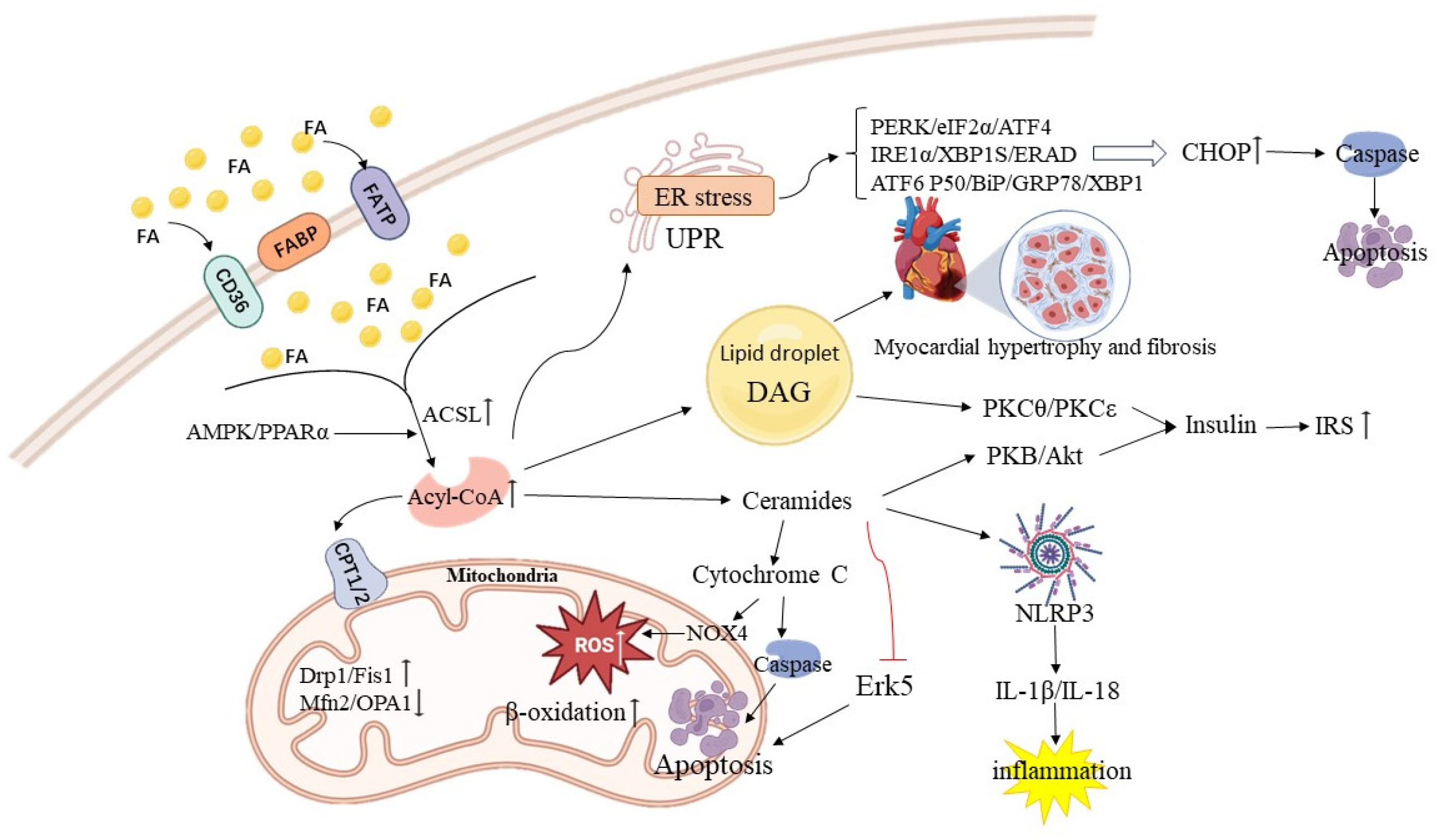
Myocardial lipotoxicity is a pathophysiological process arising from the disruption of lipid metabolic homeostasis in cardiomyocytes, particularly under metabolic disease conditions such as obesity and type 2 diabetes. Under normal physiological conditions, the heart primarily relies on fatty acids as its main energy source while maintaining metabolic flexibility [2]. However, in pathological states, excessive fatty acids are taken up by cardiomyocytes, exceeding their oxidative capacity and storage capabilities. This leads to abnormal intracellular lipid accumulation and the formation of metabolic intermediates, ultimately inducing mitochondrial dysfunction, endoplasmic reticulum stress, oxidative stress, inflammatory responses, and apoptosis, which contribute to myocardial structural remodeling and dysfunction [3,4,5].
3.1. Initial Steps in the Disruption of Myocardial Fatty Acid Metabolic Homeostasis
The uptake of fatty acids by cardiomyocytes is an active process rather than a passive diffusion, precisely regulated by various proteins. Dysregulation of this uptake is a critical initial step in the development of myocardial lipotoxicity, primarily involving abnormalities in fatty acid transport, intracellular carriers, and activation-related proteins. The FATPs and CD36 (FAT/CD36) are pivotal in mediating fatty acid uptake in cardiomyocytes. Notably, CD36 accounts for approximately 50% of long-chain fatty acid uptake in these cells, and its subcellular localization directly influences uptake efficiency [37,38]. In insulin-resistant states, although glucose uptake in cardiomyocytes diminishes, the translocation of CD36 to the cell membrane increases as a compensatory mechanism, exacerbating fatty acid uptake [39]. The Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) signaling pathway serves as a key upstream regulator of CD36 membrane localization. Under pathological conditions, abnormal AMPK activation or disruption of its downstream signaling perpetuates CD36’s anchoring to the cell membrane, creating a vicious cycle of sustained fatty acid influx [40,41]. Recent studies have highlighted that palmitoylation modification of CD36 is essential for its membrane localization and function. Inhibiting CD36 palmitoylation effectively reduces fatty acid uptake and ameliorates lipotoxicity-related myocardial metabolic disorders and dysfunction, indicating that CD36 could be a promising therapeutic target for addressing myocardial lipotoxicity [42,43]. Among the FATPs family, FATP1 and FATP6 are highly expressed in cardiac tissue, and FATP1 dysfunction is directly linked to lipotoxicity. Using animal models has shown that cardiac-specific overexpression of FATP1 can induce lipotoxic cardiomyopathy, characterized by significant myocardial lipid accumulation, cardiac hypertrophy, and heart failure [44]. FATP proteins serve dual roles: they mediate transmembrane fatty acid transport and function as very long-chain acyl-CoA synthetases that activate fatty acids transported into cells. Their excessive expression disrupts myocardial lipid homeostasis, acting as a significant trigger for lipotoxicity [45,46]. Heart-type fatty acid-binding protein (FABP3), the most abundant fatty acid carrier protein in cardiomyocytes, directs fatty acids to mitochondria (for β-oxidation), lipid droplets (for storage), or participates in signal transduction [47]. Under conditions of lipid overload, the expression and function of FABP3 are altered, leading to imbalances in the distribution of fatty acids among different metabolic pathways, further exacerbating lipid accumulation [48,49]. Simultaneously, FABP3 serves as an important biomarker of myocardial injury, with elevated circulating levels reflecting the extent of cardiomyocyte damage [50]. Fatty acids need to be activated by ACSLs to form acyl-CoA before entering oxidative or storage metabolic pathways. Among the ACSL family, ACSL1 is highly expressed in the heart, providing substrates for mitochondrial β-oxidation and raw materials for TG and complex lipid synthesis [34,51]. The expression of ACSL1 is tightly regulated by transcription factors such as PPARα. Under lipotoxic conditions, variations in ACSL activity directly influence the metabolic fate of excess fatty acids. If ACSL-mediated activation efficiency aligns with mitochondrial oxidative capacity, fatty acids can be effectively decomposed; however, if the oxidative pathway becomes saturated, excess acyl-CoA is redirected to non-oxidative metabolic pathways, laying the groundwork for the generation of toxic lipid intermediates [52,53].
3.2. Generation and Damaging Effects of Toxic Lipid Intermediates
When fatty acid influx exceeds immediate oxidative needs, a critical protective mechanism is their esterification into triglycerides (TGs) and storage within cytoplasmic lipid droplets (LDs) [54,55]. This process buffers against the accumulation of lipotoxic species like ceramides and DAG. The mobilization of this stored TG is governed by a sequential lipolytic cascade [56]. Adipose Triglyceride Lipase (ATGL), the rate-limiting enzyme, initiates lipolysis by hydrolyzing TGs to DAG, which is subsequently hydrolyzed by Hormone-Sensitive Lipase (HSL) [57]. The coordinated action of ATGL and HSL thus controls the release of fatty acids from this neutral lipid pool, determining their availability for oxidation or their potential re-routing into toxic pathways when this buffering capacity is overwhelmed [58].
Furthermore, when the fatty acid oxidation pathway in cardiomyocytes becomes saturated, excess acyl-CoA is diverted into non-oxidative metabolic pathways, leading to the production of diacylglycerol (DAG) and ceramides. These molecules are central players in myocardial lipotoxicity, exerting their toxic effects by disrupting signaling pathways and causing organelle damage. Ceramides, as key intermediates in sphingolipid metabolism, accumulate in cardiomyocytes and are closely associated with apoptosis, mitochondrial dysfunction, and insulin resistance [59,60]. Under lipotoxic conditions, ceramide synthesis occurs mainly through three pathways: (1) the de novo synthesis pathway, driven by saturated fatty acids (such as palmitic acid), where serine palmitoyltransferase (SPT) serves as the rate-limiting enzyme; (2) sphingomyelin hydrolysis, activated by inflammatory cytokines (such as TNF-α) or oxidative stress, leading to ceramide generation through the hydrolysis of cell membrane sphingomyelin; and (3) the salvage pathway, which contributes less to ceramide accumulation under pathological conditions [61]. The damaging mechanisms of ceramides encompass three main aspects: (1) inhibition of the insulin signaling pathway by directly inhibiting key protein kinase B (PKB/Akt), resulting in insulin resistance in cardiomyocytes and reduced glucose utilization [59,60]; (2) induction of oxidative stress and apoptosis through the activation of NADPH oxidase 4 (Nox4), which produces excessive reactive oxygen species (ROS) while inhibiting the protective Erk5 signaling pathway, triggering apoptotic programs [62,63]. Additionally, ceramides can directly act on mitochondria, inducing increased mitochondrial outer membrane permeability, promoting cytochrome c release, and activating caspase cascades [45,46]; (3) disruption of cell membrane microstructure: As hydrophobic molecules, ceramides can embed into the cell membrane, altering the fluidity and composition of lipid raft microdomains, interfering with receptor function and signal transduction [61]. In the Akita diabetic mouse model, elevated myocardial ceramide levels correlate directly with the development of diabetic cardiomyopathy, further underscoring their pathological significance [64,65].
DAG plays a crucial role as an intermediate in TG synthesis and functions as an important intracellular second messenger [66]. In cardiomyocytes, its accumulation primarily induces toxic effects by activating various members of the protein kinase C (PKC) family [67]. Under lipotoxic conditions, DAG—especially the sn-1,2-DAG subtype—specifically recruits and activates PKC isoforms, such as PKCθ and PKCε, at the cell membrane. Once activated, PKC disrupts normal cellular signaling by phosphorylating downstream substrates [68]. This phosphorylation of insulin receptor substrate (IRS) inhibits its tyrosine phosphorylation, effectively blocking insulin signaling and worsening myocardial insulin resistance [69]. Moreover, aberrant PKC activation is closely linked to myocardial hypertrophy, fibrosis, and contractile dysfunction [70]. Diacylglycerol kinase (DGK) is a critical enzyme that regulates cellular DAG levels by catalyzing the conversion of DAG to phosphatidic acid, thereby decreasing intracellular DAG concentrations. While the exact mechanisms by which DGK influences myocardial lipotoxicity require further exploration, targeted modulation of DGK activity to manage the DAG-PKC signaling pathway has emerged as a promising therapeutic approach for mitigating myocardial lipotoxicity [71].
3.3. Organelle Dysfunction Mediated by Lipotoxicity
The buildup of toxic lipids, such as ceramides and DAG, significantly disrupts the functional homeostasis of key organelles within cardiomyocytes, with the mitochondria and the endoplasmic reticulum being the primary targets [6]. These organelles’ stress responses exacerbate lipotoxic damage. Mitochondria, which are essential for fatty acid β-oxidation, are particularly susceptible to lipotoxic injury [72]. Their dysfunction is primarily marked by increased oxidative stress, impaired energy metabolism, and a disrupted dynamic balance [73]. When fatty acid influx into mitochondria exceeds the capacity of the β-oxidation pathway, it results in an overproduction of ROS by the electron transport chain (ETC). These ROS inflict damage on mitochondrial DNA, structural proteins, and membrane lipids, creating a detrimental cycle of “oxidative damage–functional decline” [2,74]. To manage excess energy, mitochondrial uncoupling proteins (UCPs) in the inner membrane are activated [75]. By uncoupling the proton gradient from ATP synthesis, UCPs dissipate energy as heat. While this compensatory mechanism may temporarily reduce ROS production, chronic activation ultimately impairs ATP synthesis efficiency, leading to an energy crisis in cardiomyocytes [76]. Mitochondria maintain their morphology, quantity, and functional integrity through continuous fission and fusion processes, which are severely disrupted under conditions of myocardial lipotoxicity [77]. Notably, key proteins that mediate mitochondrial fission, such as dynamin-related protein 1 (Drp1) and its receptor Fis1, are significantly upregulated. This excessive fission results in fragmentation of the mitochondrial network, producing small, spherical mitochondria with compromised function. These dysfunctional organelles are either subjected to autophagic clearance or release pro-apoptotic factors [78,79]. Animal studies have demonstrated that pharmacological inhibition of Drp1-mediated fission can mitigate myocardial injury induced by ischemia-reperfusion [80]. Conversely, the expression of mitochondrial fusion proteins, including Mitofusin 2 (Mfn2) for outer membrane fusion and optic atrophy 1 (OPA1) for inner membrane fusion, is downregulated. OPA1 is crucial for maintaining cristae structure and the stability of respiratory chain complexes. Reduced expression or abnormal processing of OPA1 (as evidenced by an altered l-OPA1/S-OPA1 ratio) severely compromises mitochondrial fusion capacity, leading to diminished ATP production and reduced cell survival [81]. Consequently, promoting mitochondrial dynamics—such as enhancing fusion and inhibiting excessive fission—has emerged as a promising therapeutic strategy for treating cardiomyopathy.
The endoplasmic reticulum serves as the central site for protein synthesis, folding, and lipid synthesis [82]. An overload of lipids disrupts endoplasmic reticulum homeostasis, resulting in the accumulation of unfolded or misfolded proteins within the lumen, which triggers endoplasmic reticulum stress and the unfolded protein response (UPR) [83,84]. The UPR activates adaptive responses through three parallel signaling pathways: PERK, IRE1α, and ATF6 [85]. However, if stress persists, the adaptive response shifts toward pro-apoptotic signals, further aggravating myocardial damage. Upon activation, protein kinase R-like endoplasmic reticulum kinase (PERK) phosphorylates eukaryotic initiation factor 2α (Eif2α), inhibiting global protein translation to alleviate the endoplasmic reticulum folding burden [86]. Concurrently, the phosphorylation of Eif2α selectively promotes the translation of transcription factor ATF4 [87]. ATF4 translocates to the nucleus, where it upregulates the expression of apoptosis-related genes, particularly CHOP/C/EBP homologous protein, which is a key executor of endoplasmic reticulum stress-induced apoptosis [88]. Inositol-requiring protein 1α (IRE1α), the most conserved sensor in the UPR, activates its endonuclease activity upon activation, splicing X-box binding protein 1 (XBP1) mRNA to produce the active transcription factor XBP1s. XBP1s upregulates genes associated with protein folding, transport, and endoplasmic reticulum associated degradation (ERAD), helping restore endoplasmic reticulum homeostasis [89,90,91]. However, under prolonged stress, IRE1α activates pro-apoptotic signals such as c-Jun N-terminal kinase (JNK), further damaging cells. Activating transcription factor 6 (ATF6) moves from the endoplasmic reticulum membrane to the Golgi apparatus under stress conditions, where it is cleaved by proteases to release its active cytoplasmic domain (ATF6 p50). This domain enters the nucleus to upregulate genes for chaperone proteins, including BiP/GRP78, and XBP1, enhancing the ER’s folding capacity [92]. Under persistent lipotoxic stress, all three UPR pathways significantly increase CHOP expression, ultimately activating caspase-12 (in mice) or caspase-4 (in humans) and downstream caspase-3, initiating the apoptotic program [93].
3.4. Synergistic Damaging Effects of Inflammatory Response and Programmed Cell Death
Lipotoxicity not only disrupts intracellular metabolism and organelle function but also triggers systemic inflammatory responses and various forms of programmed cell death, resulting in significant cardiomyocyte loss and exacerbating cardiac structural remodeling and functional deterioration [6,62]. Excessive saturated fatty acids and ceramides can act as “damage-associated molecular patterns” (DAMPs), activating the NLRP3 inflammasome in cardiomyocytes [94]. Once assembled, the NLRP3 inflammasome activates caspase-1, leading to the cleavage and activation of interleukin-1β (IL-1β) and IL-18. These pro-inflammatory cytokines recruit immune cells, including monocytes and macrophages, to myocardial tissue, intensifying local inflammation and tissue damage, thereby creating a positive feedback loop of “inflammation-lipotoxicity” [95,96]. Myocardial lipotoxicity can induce various forms of programmed cell death, collectively contributing to cardiomyocyte loss: (1) Apoptosis: As previously mentioned, the mitochondrial pathway (cytochrome c release) and the endoplasmic reticulum stress pathway (CHOP upregulation) are the primary mechanisms of lipotoxicity-induced apoptosis [7]; (2) Necroptosis and Pyroptosis: Recent studies have confirmed that these two forms of programmed necrosis also play a role in lipotoxic myocardial injury. Pyroptosis is directly linked to NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activation, while necroptosis occurs through the RIPK1/RIPK3/MLKL signaling axis [97,98]; (3) Lipid accumulation-related death: Under hypoxic or ischemic conditions, TG accumulation in cardiomyocytes can lead to endoplasmic reticulum stress, mitochondrial dysfunction, and inflammatory responses, further promoting cell death [30]. Additionally, abnormal expression of specific proteins can exacerbate lipotoxic damage; for instance, the very low-density lipoprotein receptor (VLDLR) facilitates lipid accumulation in HL-1 cardiomyocytes and mouse hearts under hypoxic or ischemic conditions [80]. Cardiomyocyte-specific overexpression of FATP1 or ACSL1 increases the uptake of FFAs, leading to the accumulation of toxic lipids such as ceramides and TGs. This, in turn, activates iNOS, pro-hypertrophic signals, and apoptotic pathways, ultimately resulting in heart failure [99]. To fully appreciate the supply side of lipid overload, it is crucial to consider systemic and local regulatory mechanisms. The adipose-heart axis plays a significant role, where dysfunctional adipose tissue in obesity releases excessive fatty acids and pro-inflammatory adipokines into the circulation, directly contributing to the lipid burden faced by the heart [100]. Furthermore, at the coronary endothelium, the processing of triglyceride-rich lipoproteins is a critical step. Lipoprotein lipase (LPL), the key enzyme hydrolyzing triglycerides into fatty acids for cardiac uptake, is anchored to the capillary lumen by its transporter, glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1). The transcytosis of the LPL–GPIHBP1 complex across endothelial cells ensures its proper positioning and function, and disruptions in this process can significantly impact myocardial lipid delivery [101]. These studies suggest that the fundamental issue of myocardial lipotoxicity lies in the imbalance between lipid supply and oxidative capacity, culminating in the accumulation of lipotoxic substances, rather than merely an increase in fatty acids intake (Figure 2).
4. Impact of Myocardial Lipotoxicity on Cardiovascular Diseases
4.1. Heart Failure
Heart failure, a severe complication of impaired myocardial lipid metabolism, is intricately linked to cardiac energy metabolic remodeling and the resulting cascade of lipotoxic injury [8,92]. In a healthy heart, around 60–70% of ATP is produced through fatty acid β-oxidation (FAO) [1]. However, during heart failure, myocardial energy metabolism experiences profound reprogramming. Initially, as a compensatory response to hypoxia and oxidative stress, the heart transitions from primarily utilizing fatty acids to increased glucose reliance. As the condition progresses, this metabolic shift becomes maladaptive and ultimately leads to decompensation [102]. This phase is characterized by persistently diminished fatty acid oxidation capacity, a compensatory increase in glycolysis, and impaired glucose oxidation due to inadequate pyruvate entry into mitochondria, resulting in significantly reduced energy production efficiency [103]. This metabolic disruption not only creates an “energy-starved” state but also disturbs lipid homeostasis, paving the way for lipotoxicity. In metabolic disorders like obesity and type 2 diabetes, elevated circulating FFAs are excessively absorbed by cardiomyocytes via the fatty acid transporter CD36 [36,104]. Simultaneously, mitochondrial dysfunction and reduced activity of crucial FAO enzymes (e.g., CPT-Ⅰ) create a severe imbalance between fatty acid influx and oxidation [105,106]. Excess fatty acids are rerouted into non-oxidative pathways, resulting in cytoplasmic storage as TG or conversion into harmful lipid intermediates like DAG and ceramides. These toxic lipids disrupt critical signaling pathways, inflicting multifaceted damage on cardiomyocyte structure and function [107,108].
Ceramides and DAG serve as pivotal effector molecules in mediating myocardial lipotoxicity, intricately regulating signaling pathways that trigger apoptosis, metabolic dysregulation, and organelle dysfunction. Ceramide, a crucial signaling molecule within sphingolipid metabolism, contributes to cellular injury through three primary mechanisms [109,110]. First, it directly activates caspase cascades, facilitates cytochrome c release through mitochondrial outer membrane permeabilization, and inhibits pro-survival AKT signaling, ultimately inducing apoptosis. Notably, inhibiting ceramide synthesis significantly diminishes apoptosis [111,112]. Second, ceramide activates protein phosphatase 2A (PP2A) and atypical protein kinase Cζ (PKCζ), which inhibit the tyrosine phosphorylation of insulin receptor substrate 1 (IRS-1), thereby impairing AKT activation. This disruption in turn hinders the membrane translocation of glucose transporter 4 (GLUT4), exacerbating the energy crisis and diminishing AKT-mediated anti-apoptotic effects [113,114,115,116]. Third, ceramide interferes with endoplasmic reticulum calcium homeostasis, triggering the UPR. Sustained endoplasmic reticulum stress leads to an upregulation of CHOP (C/EBP homologous protein) and activation of caspase-12, promoting apoptosis [72]. In failing hearts, markers of endoplasmic reticulum stress (e.g., GRP78, CHOP) show a positive correlation with ceramide levels, and the endoplasmic reticulum stress inhibitor 4-phenylbutyric acid (4-PBA) has been shown to improve cardiac function [83]. Meanwhile, DAG primarily exerts its toxicity by activating specific PKC isoforms [117]. PKCθ phosphorylates IRS-1 at serine 307, thereby blocking its interaction with the insulin receptor, inhibiting AKT activation, and impairing GLUT4 translocation [67,118]. In cases of cardiac dysfunction induced by a high-fat diet, the knockout of PKCθ mitigates DAG-induced damage [119]. Additionally, PKCδ activates pro-hypertrophic transcription factors like NF-κB, promoting long-term maladaptive hypertrophy and eventual decompensation [30]. Moreover, PKC-mediated phosphorylation of ryanodine receptor 2 (RyR2) and phospholamban (PLN) leads to calcium leakage from the sarcoplasmic reticulum (SR) and enhances the inhibition of SR Ca2+-ATPase (SERCA2a), ultimately impairing diastolic function and reducing contractility [120,121].
Mitochondria, as the primary site of fatty acid β-oxidation, are critical targets for lipotoxicity. Mitochondrial dysfunction directly contributes to energy depletion in heart failure, a process carefully regulated by the AMPK/PGC-1α/PPARα signaling axis. Excess lipids cause the uncoupling of the mitochondrial electron transport chain, leading to the overproduction of ROS[115]. ROS can damage mitochondrial DNA, respiratory chain complexes, and mitochondrial membranes, creating a vicious cycle of “ROS generation–mitochondrial damage–further ROS production” [107,108,109,112]. This results in reduced efficiency of oxidative phosphorylation and a sharp decline in ATP synthesis, which is insufficient to support cardiac contraction and relaxation [122]. Lipotoxicity also disrupts mitochondrial dynamics by downregulating fusion proteins (Mfn1/2, OPA1) and upregulating fission proteins (Drp1, Fis1), leading to mitochondrial fragmentation. This further impairs energy production and increases the risk of apoptotic factor release [107,123]. The dysregulation of the AMPK/PGC-1α/PPARα axis exacerbates mitochondrial dysfunction. PPARα, the master regulator of fatty acid metabolism, controls the expression of genes related to FAO, such as CD36 and CPT1 [104,124]. In heart failure, its expression and activity are significantly diminished [36]. PGC-1α, a coactivator of PPARα, regulates both FAO and mitochondrial biogenesis; its impairment compromises energy substrate utilization and mitochondrial capacity simultaneously [103]. AMPK, functioning as a cellular energy sensor, is typically activated under energy-deficient conditions to promote the PGC1α–PPARα pathway [125]. However, in advanced heart failure, AMPK activation is blunted, failing to initiate necessary compensatory mechanisms [103]. In a mouse model of heart failure with preserved ejection fraction (HFpEF) supplementation with astragaloside IV has been shown to activate the Nrf2/HO-1 signaling pathway, resulting in the upregulation of antioxidant enzymes (e.g., HO-1, NQO-1) and modulation of the Bcl-2/Bax balance, which attenuates oxidative stress and improves cardiac function [126]. These insights underscore the potential of targeting mitochondrial function to counteract myocardial lipotoxicity (Table 1).
| Core Component | Key Pathological Changes/Molecules | Specific Mechanisms and Consequences | Functional Impact | References |
|---|---|---|---|---|
| 1. Metabolic Remodeling & Lipid Accumulation | Metabolic Reprogramming |
Impaired Fatty Acid Oxidation (FAO): Reduced activity of key enzymes (e.g., CPT1) and mitochondrial dysfunction. Dysregulated Glucose Utilization: Compensatory increase in glycolysis, but impaired glucose oxidation (blocked pyruvate entry into mitochondria). Result: Significantly reduced ATP production efficiency, leading to myocardial “energy starvation.” |
Inadequate energy supply, impairing the basis of cardiac contraction and relaxation. | [102,103] |
| Disruption of Lipid Homeostasis |
Background: Elevated circulating FFAs due to metabolic syndromes (e.g., diabetes). Process: Excessive FFA uptake via CD36, coupled with insufficient oxidative capacity, results in lipid “influx > efflux.” Result: Excess fatty acids are diverted into toxic lipid species like DAG and ceramide. |
Establishes the foundation for lipotoxic injury. | [36,104,105,106,107,108] | |
| 2. Lipotoxic Injury (Core Effector Molecules) | Ceramide |
1. Induces Apoptosis: Activates caspase cascade, promotes cytochrome c release, and inhibits pro-survival Akt signaling. 2. Causes Insulin Resistance: Activates PP2A/PKCζ, inhibits IRS-1 tyrosine phosphorylation and Akt activation, impedes GLUT4 translocation, worsening energy crisis. 3. Triggers ER Stress: Disrupts calcium homeostasis, induces the UPR, and initiates apoptosis via CHOP/caspase-12 upregulation. |
Cardiomyocyte death, metabolic dysregulation, and organelle dysfunction. | [49,72,83,109,110,111,112,113,114,115,116] |
| DAG |
1. Causes Insulin Resistance: Activates PKCθ, leading to IRS-1 phosphorylation (Ser307), blunting insulin signaling. 2. Promotes Hypertrophy: activates PKCδ, which subsequently activates pro-hypertrophic transcription factors like NF-κB. 3. Disrupts calcium handling: PKC activation phosphorylates RyR2 (causing calcium leak) and PLN (enhancing inhibition of SERCA2a). |
Insulin resistance, cardiac hypertrophy, and diastolic/systolic dysfunction. | [69,117,118,119,120,121] | |
| 3. Mitochondrial dysfunction | Oxidative stress & damage |
ROS Burst: lipid overload causes electron transport chain uncoupling, generating excessive ROS.
Vicious Cycle: ROS damages mitochondrial components (DNA, proteins, membranes), creating a “ROS-induced damage-increased ROS” cycle. Result: decreased oxidative phosphorylation efficiency and a sharp decline in ATP synthesis. |
Direct cause of energy failure. | [107,108,109,115,122] |
| Dynamical imbalance |
Fragmentation: downregulation of fusion proteins (Mfn1/2, OPA1) and upregulation of fission proteins (Drp1, Fis1). Result: fragmented mitochondrial network, reduced functional capacity, and increased risk of pro-apoptotic factor release. |
Further compromises energy production capacity. | [107,123] | |
| 4. Dysregulation of key signaling axis | AMPK/PGC-1α/PPARα Axis |
PPARα↓: downregulation and inactivation of this master regulator of fatty acid metabolism reduces CD36, CPT1, etc. PGC-1α↓: impaired function of this coactivator disrupts both FAO and mitochondrial biogenesis. AMPK↓: blunted activation of this cellular energy sensor fails to initiate compensatory pathways during energy deficit. Result: exacerbates mitochondrial dysfunction and metabolic imbalance. |
Loss of core regulatory control over metabolic homeostasis, accelerating HF progression. | [36,103,104,124,125] |
| 5. Potential therapeutic directions | Nrf2/HO-1 pathway | In HFpEF models, activating this pathway upregulates antioxidant enzymes (e.g., HO-1, NQO-1), modulates apoptotic balance (Bcl-2/Bax), and alleviates oxidative stress. | Indicates antioxidant therapy as a promising strategy. | [126] |
4.2. Arrhythmias
Myocardial lipotoxicity leads to the abnormal accumulation of bioactive lipid intermediates in cardiomyocytes, forming a complex pathological network referred to as “lipo-arrhythmogenesis.” This condition arises through direct modulation of ion channel function, disruption of calcium homeostasis, induction of oxidative stress, and remodeling of electrical conduction pathways [9]. The primary mechanisms involve three interrelated aspects: abnormal triggered activity, calcium dysregulation, and impaired electrical conduction [127]. Regarding triggered activity, ceramide—a key bioactive sphingolipid—impairs the rapidly activating delayed rectifier potassium current (IKr) through two main pathways: direct binding to the hERG channel protein and activation of protein phosphatases (PP1/PP2A), leading to channel dephosphorylation, reduced function, and decreased membrane expression [128]. The inhibition of IKr significantly prolongs action potential duration (APD) and the effective refractory period (ERP), increasing the likelihood of l-type calcium channel (LTCC) recovery from inactivation and subsequent reopening, which can induce early afterdepolarizations (EADs) [129]. These EADs serve as triggers for malignant ventricular arrhythmias, such as Torsades de Pointes (TdP). Long-chain acylcarnitines (LCACs), which are intermediate metabolites of fatty acid β-oxidation, accumulate intracellularly during metabolic stress due to an imbalance between production and clearance. Their amphiphilic properties enable them to integrate into cell membranes and the lipid microenvironment of ion channels, leading to a dose-dependent inhibition of IKr, sodium current (INa), and L-type calcium current (ICa-L) [130]. By delaying repolarization, LCACs extend APD and promote EADs. Additionally, LCACs enhance the opening of RyR2, increasing diastolic calcium leakage and creating conditions conducive to delayed afterdepolarizations (DADs), resulting in a synergistic pro-arrhythmic effect [131]. Notably, LCACs exhibit a concentration-dependent dual effect on potassium channels: at low concentrations, they activate hERG channels and shorten APD, while at high concentrations, they inhibit the inward rectifier potassium current (IK1) and ATP-sensitive potassium channels (IKATP), thereby slowing repolarization [4,130]. Both scenarios ultimately compromise myocardial repolarization reserve, heightening the susceptibility to arrhythmias.
Oxidative stress-driven disruption of calcium homeostasis serves as a key amplifying mechanism by which lipid metabolic disorders contribute to arrhythmias. Concurrently, chronic inflammation-induced electrical remodeling establishes the structural foundation for re-entrant arrhythmias [132]. In lipotoxic environments, ROS target polyunsaturated fatty acids, leading to lipid peroxidation and the formation of reactive aldehydes like 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) [95]. These aldehydes covalently modify (carbonylate) essential calcium-handling proteins, thereby impairing their functionality [133]. Specifically, 4-HNE alters thiol groups on RyR2, which increases its open probability and causes diastolic calcium leakage from the SR [134]. This leakage depletes SR calcium stores, weakens systolic calcium transients, and results in cytosolic calcium overload [135]. At the same time, 4-HNE inhibits the activity of SERCA2a, thereby slowing diastolic calcium reuptake and exacerbating calcium dysregulation [136]. The rise in cytosolic calcium enhances the forward-mode activity of the sodium-calcium exchanger (NCX), which exchanges one Ca²⁺ for three Na⁺ ions, resulting in a net inward current (INCX) that underlies DADs. If DADs reach threshold potential, they may trigger tachyarrhythmias such as ventricular tachycardia [137,138]. Regarding electrical conduction remodeling, chronic low-grade inflammation linked to disordered lipid metabolism alters cardiomyocyte electrical coupling by affecting connexin 43 (Cx43) [139]. Reduced adiponectin levels lead to diminished activation of the AMPK signaling pathway—a critical regulator of Cx43 phosphorylation—while elevated leptin levels promote myocardial fibrosis through the JAK2/STAT3 pathway, which isolates cardiomyocytes. Additionally, inflammatory cytokines like tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) activate the NF-κB and p38 MAPK/ERK1/2 pathways, resulting in decreased total Cx43 protein expression, promoting its dephosphorylation, and causing its lateralization from intercalated discs to lateral cell membranes [140]. The reduction, functional impairment, and abnormal distribution of Cx43 slow conduction velocity and modify conduction anisotropy, thereby facilitating functional conduction block and creating a substrate for reentry—fundamental mechanisms sustaining arrhythmias such as atrial and ventricular fibrillation [141].
In summary, disrupted myocardial lipid metabolism fosters arrhythmogenesis through a multifaceted network that includes: (1) direct modulation of ion channels by bioactive lipids; (2) amplification of oxidative stress-induced calcium dysregulation; and (3) structural remodeling of electrical conduction due to chronic inflammation. These pathways interact in positive feedback loops, such as calcium overload further promoting ROS generation and inflammation exacerbating abnormal lipid accumulation. Future therapeutic approaches for metabolic arrhythmias should move beyond traditional ion channel blockade to focus on “upstream” regulation of lipid metabolism. Strategies may include targeted clearance of toxic lipids (e.g., ceramides and LCACs), activation of AMPK to enhance Cx43 function, and inhibition of NF-κB to mitigate inflammatory responses. Such interventions could open new avenues for preventing and treating arrhythmias linked to lipotoxicity at their source (Table 2).
| Core Mechanism | Key Mediators/Factors | Targets/Pathways Affected | Electrophysiological Consequences & Arrhythmogenic Effects | References |
|---|---|---|---|---|
| 1. Abnormal triggered activity | Ceramide |
Direct hERG channel inhibition: binding to the channel protein. Indirect inhibition: activation of protein phosphatases PP1/PP2A, leading to channel dephosphorylation. |
Prolonged APD and increased ERP. Increased probability of L-type calcium channel re-opening, inducing EADs, which can initiate tachyarrhythmias like TdP. |
[128,129,130] |
| LCACs |
Non-specific inhibition of multiple ion channels: dose-dependent inhibition of IKr, INa, and ICa-L. RyR2 activation: promoting diastolic calcium leak from the SR. |
Inhibition of repolarizing currents prolongs APD and induces EADs. SR calcium leak causes cytosolic calcium overload, promoting DADs, synergizing with EADs. Impairment of repolarization reserve, increasing arrhythmia susceptibility. |
[130,131] | |
| 2. Calcium homeostasis dysregulation | ROS & reactive aldehydes (e.g.,4-HNE, MDA) |
RyR2 modification: Increased open probability, leading to diastolic SR calcium leak. SERCA2a inhibition: impaired diastolic calcium reuptake, exacerbating calcium overload. |
Elevated cytosolic calcium concentration.
Activating the forward mode of the NCX generates a net INCX. INCX serves as the direct basis for the formation of DADs and can trigger rapid arrhythmias. |
[134,135,136,137,138] |
| 3. Electrical conduction disturbance | Chronic Low-Grade inflammation |
Adiponectin↓/Leptin↑: resulting in suppressed AMPK signaling (reduced Cx43 phosphorylation) and activation of the JAK2/STAT3 pathway (promoting fibrosis). Inflammatory cytokines (TNF-α, IL-1β): activation of NF-κB and p38 MAPK/ERK pathways, leading to downregulation of total Cx43 expression, its dephosphorylation, and lateralization. |
Impaired gap junction function and weakened intercellular electrical coupling. Slowed conduction velocity and increased conduction anisotropy. Creation of a substrate for functional conduction block and re-entry, sustaining reentrant arrhythmias (e.g., atrial fibrillation, ventricular fibrillation). |
[139,140,141] |
4.3. Atherosclerosis
Atherosclerosis development hinges on the “lipid infiltration theory”, which posits that the pathological process begins with the accumulation of oxidized low-density lipoprotein (oxLDL) due to disordered lipid metabolism [10]. This process unfolds through a sequence involving various cells and signaling pathways, ultimately resulting in endothelial dysfunction, foam cell formation, chronic inflammation, and plaque destabilization [142]. When circulating low-density lipoprotein cholesterol (LDL-C) levels rise, LDL particles infiltrate the compromised arterial endothelium into the subendothelial space, where they are modified into oxLDL by local ROS. Acting as a pivotal bioactive molecule, oxLDL instigates pathological signaling primarily through the lectin-like oxidized LDL receptor-1 (LOX-1) on endothelial cells [143,144]. Activation of LOX-1 inhibits endothelial nitric oxide synthase (eNOS) activity via PKC and MAPK pathways, leading to decreased production of the vasodilator nitric oxide (NO) and resulting in vasomotor dysfunction [145]. Concurrently, LOX-1-mediated uptake of oxLDL strongly activates the nuclear factor-kappa B (NF-κB) pathway, prompting endothelial cells to release pro-inflammatory cytokines such as interleukin-8 (IL-8) and tumor necrosis factor-alpha (TNF-α), while also upregulating vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin to facilitate monocyte adhesion and migration [146]. In this context, microRNAs like miR-49 and let-7 g can inhibit LOX-1 expression by targeting its mRNA, thereby exerting protective effects [147,148]. This shift in endothelial cell function from “guardians” to “pro-inflammatory promoters” signals the onset of early atherosclerotic progression.
Upon entering the subendothelial space, monocytes differentiate into macrophages. They take up oxLDL uncontrollably through scavenger receptors (SR-A and CD36), bypassing intracellular cholesterol’s negative feedback regulation, and eventually transform into foam cells, which are the primary constituents of the plaque lipid core [149,150]. Additionally, oxLDL functions as a damage-associated molecular pattern (DAMP), that activates Toll-like receptor 4 (TLR4) on macrophages, triggering the MyD88-dependent pathway, which activates NF-κB and MAPK signaling, thereby intensifying inflammation. Moreover, oxLDL interacts with the mTORC2-Akt-mTORC1 metabolic axis [151,152]. Activation of the PI3K/Akt pathway promotes oxLDL uptake, while subsequent mTOR activation inhibits SIRT1’s deacetylase activity, enhancing lipid synthesis and uptake while suppressing cholesterol efflux [2,153], thereby creating a vicious cycle of lipid accumulation [154,155,156]. MicroRNAs such as hsa-miR-758-5p and hsa-miR-204-3p can downregulate CD36 expression by targeting its mRNA, which slows foam cell formation [157]. The precipitation of supersaturated cholesterol into cholesterol crystals (CCs) within foam cells, coupled with oxLDL-induced mitochondrial dysfunction (characterized by disrupted electron transport and increased mitochondrial ROS production), collectively triggers NLRP3 inflammasome activation [158]. Accumulation of impaired mitochondria results from inadequate mitophagy, which further exacerbates mitochondrial ROS release and cytochrome c leakage, ultimately facilitating the assembly of NLRP3 with ASC and pro-caspase-1 into an active inflammasome [159,160,161,162]. Activated caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature pro-inflammatory forms and also cleaves Gasdermin D, inducing pyroptosis in foam cells and releasing large amounts of lipids and inflammatory contents, thereby establishing a positive feedback loop of “inflammation-lipid accumulation” [163].
The phenotypic switching of vascular smooth muscle cells (VSMCs) is a pivotal event in the transition of atherosclerosis from early fatty streaks to advanced plaques, driven by persistent metabolic regulatory network imbalances throughout the pathology. Normally quiescent “contractile” VSMCs are stimulated in the plaque microenvironment by platelet-derived growth factor (PDGF), secreted by macrophages and endothelial cells. Through the PDGFR-β/FAK pathway, these cells initiate migration and proliferation, synthesizing and secreting extracellular matrix components to form the plaque fibrous cap [164,165]. In advanced lesions, Kruppel-like factor 4 (KLF4) and Oct4 collaborate to drive VSMCs toward unstable phenotypes resembling macrophages and osteoblasts, which contribute to plaque calcification and fibrous cap thinning. Additionally, the Hippo pathway effectors YAP/TAZ become activated under disturbed flow and inflammatory stimuli, promoting VSMC proliferation and vascular remodeling, which further destabilizing plaques [166]. Throughout this process, metabolic regulation is governed by the liver X receptor (LXR)/peroxisome proliferator-activated receptor (PPAR) and AMPK/mTOR signaling axes. LXR (activated by oxidized cholesterol) and PPAR (activated by fatty acids) upregulate ATP-binding cassette transporters A1/G1 (ABCA1/ABCG1), facilitating reverse cholesterol transport while also inhibiting the NF-κB pathway to exert anti-inflammatory effects [167,168,169]. Under conditions of energy deficiency, AMPK activation inhibits mTORC1, promoting autophagy, clearing damaged mitochondria, suppressing NLRP3 activation [170], and enhancing LXR/PPAR activity. However, in hyperlipidemic and pro-inflammatory environments, mTOR is overactivated and AMPK is suppressed, leading to a disruption of metabolic homeostasis that exacerbates lipid accumulation, inflammation, and abnormal VSMC proliferation [171].
In summary, the development and progression of atherosclerosis are driven by disordered lipid metabolism through a complex cascade involving multiple stages, cell types, and pathways. The process initiates when oxLDL attacks the vascular endothelium, compromising its barrier function by activating pathways such as NF-κB. Subsequently, macrophages excessively phagocytose lipids via scavenger receptors and signaling pathways, including TLR4/mTOR, leading to their transformation into foam cells. Within these foam cells, danger signals such as cholesterol crystals and mtROS trigger a vicious cycle of NLRP3 inflammasome activation and impaired mitophagy, perpetuating inflammatory responses. Ultimately, VSMCs undergo phenotypic switching under the influence of signals such as PDGF, KLF4, and YAP/TAZ, resulting in plaque growth, thinning of the fibrous cap, and destabilization of the atherosclerotic lesions. This entire process is underpinned by an imbalance in metabolic regulatory networks centered around LXR/PPAR and AMPK/mTOR. A comprehensive understanding of these intricate molecular mechanisms and the interplay between signaling pathways offers a robust theoretical foundation and identifies potential targets for innovative strategies aimed at preventing and treating atherosclerosis by intervening at specific points in this cascade (Table 3).
| Pathological Stage | Key Cellular Event | Critical Molecules/Pathways | Specific Actions and Consequences | References |
|---|---|---|---|---|
| 1. Endothelial dysfunction | Lipid infiltration and endothelial activation LDL enters the subendothelial space, becomes oxidized to oxLDL, and activates endothelial cells. | oxLDL/LOX-1 receptor |
Inhibits eNOS activity: reduces NO production via the PKC/MAPK pathway, causing vasodilatory dysfunction. Activates NF-κB pathway: induces expression of adhesion molecules (VCAM-1/ICAM-1) and secretion of pro-inflammatory cytokines (TNF-α/IL-8), recruiting monocytes. Protective miRNAs: miRNAs like miR-49 and let-7 g can target and suppress LOX-1 expression, exerting a protective effect. |
[10,142,143,144,145,146,147,148] |
| 2. Foam cell formation | Monocyte migration into the intima, differentiation into macrophages, and unregulated uptake of oxLDL to form foam cells. | Scavenger receptors (SR-A, CD36) |
Unregulated lipid uptake: leads to massive intracellular lipid accumulation, forming the plaque’s lipid core. Protective miRNAs: miRNAs like hsa-miR-758-5p can target CD36, slowing foam cell formation. |
[149,150] |
| Inflammation amplification and metabolic dysregulation | TLR4/NF-κB pathway & mTORC2-Akt-mTORC1 axis |
Amplifies inflammation: oxLDL acts as a DAMP to activate TLR4, amplifying inflammation via the NF-κB/MAPK pathway. Promotes lipid accumulation: Akt/mTOR signaling activation inhibits SIRT1, simultaneously promoting lipid uptake and inhibiting cholesterol efflux, creating a vicious cycle. |
[2,151,152,153,154,155,156] | |
| 3. Plaque progression & destabilization | Inflammasome activation and pyroptosis cholesterol crystals and mtROS trigger intense inflammatory responses. | NLRP3 Inflammasome |
Triggering activation: activated by cholesterol crystals and mitochondrial dysfunction (mtROS, impaired mitophagy). Cytokine release: activated caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature, active forms. Induces pyroptosis: cleaves gasdermin D, leading to macrophage pyroptosis and the release of inflammatory contents, worsening local inflammation and necrotic core formation. |
[159,160,161,162,163] |
| VSMCs transition from a contractile to a synthetic/macrophage-like phenotype. | PDGF/KLF4&Oct4 YAP&TAZ |
Migration and proliferation: PDGF, via the PDGFR-β/FAK pathway, drives VSMC migration into the intima and proliferation, contributing to the fibrous cap. Dedifferentiation: KLF4 and Oct4 drive VSMCs toward unstable phenotypes, weakening the fibrous cap and promoting calcification. Mechanosensing: YAP/TAZ activation promotes VSMC proliferation and vascular remodeling, contributing to plaque instability. |
[164,165,166] | |
| 4. Overarching metabolic regulation | Cholesterol reverse transport and Anti-inflammation | LXR/PPAR Pathways |
Promotes cholesterol efflux: upon activation, they upregulate ABCA1/ABCG1, facilitating cholesterol export to ApoA-I/HDL. Exerts anti-inflammatory effects: suppresses the NF-κB pathway. |
[167,168,169,170,171] |
5. Therapeutic Strategies and Future Research Directions
5.1. Current Therapeutic Strategies
Current therapeutic approaches to myocardial lipotoxicity primarily focus on modulating lipid metabolism, alleviating cardiac load, and intervening in specific molecular pathways. Lifestyle interventions form the cornerstone of treatment, emphasizing dietary changes and physical activity to reduce lipid intake and enhance lipid utilization. Research indicates that moderate weight loss can markedly decrease myocardial lipid content, thereby improving cardiac function. In terms of pharmacological treatments, various cardiovascular drugs have shown promise in mitigating myocardial lipotoxicity, yet their limitations warrant consideration. SGLT2 inhibitors, such as empagliflozin and dapagliflozin, have been validated in multiple large randomized controlled trials to significantly lower the incidence of cardiovascular events and cardiovascular-related mortality among patients with heart failure, including those with and without diabetes [172,173]. The cardioprotective mechanisms of these agents are multifaceted, including promoting urinary glucose excretion, optimizing energy metabolism, and inhibiting inflammatory and fibrotic processes. However, their efficacy can be influenced by renal function, and side effects such as genitourinary infections and euglycemic diabetic ketoacidosis, though rare, require clinical vigilance [174].
Trimetazidine, a metabolic modulator, has been shown to enhance cardiac function by shifting myocardial energy metabolism from fatty acid oxidation to glucose oxidation [175,176,177]. Numerous randomized controlled trials and meta-analyses have demonstrated that trimetazidine can improve left ventricular ejection fraction in heart failure patients, enhance NYHA functional classification, boost exercise capacity, and reduce rehospitalization rates [178]. Nevertheless, its class-specific side effects, such as parkinsonism and other gait disorders, have led to usage restrictions in some countries, highlighting a risk-benefit consideration that may limit its long-term use in certain populations [179].
PPARα agonists, such as fibrates, activate the PPARα transcription factor, regulate the expression of lipid metabolism-related genes, and lower serum triglyceride levels [180,181]. Nonetheless, the cardiovascular benefits of these drugs remain contentious; for instance, the selective PPARα modulator pemafibrate did not achieve a significant reduction in cardiovascular events in the PROMINENT trial [182,183]. Furthermore, older fibrates like gemfibrozil carry a risk of myopathy and rhabdomyolysis, particularly when co-administered with statins, and can elevate serum creatinine levels [184].
5.2. Emerging Therapeutic Strategies and Future Directions
Future therapeutic strategies for myocardial lipotoxicity should place greater emphasis on targeting specific molecular pathways, developing personalized treatment plans, and addressing fundamental scientific challenges. ACC inhibitors act directly on acetyl-CoA carboxylase (ACC), a key enzyme in de novo lipogenesis, thereby reducing the production of toxic lipid species [185]. Preclinical studies have indicated that ACC2 knockout mice display protection against cardiac hypertrophy [186]. However, there is a lack of robust evidence from human clinical trials. Therapeutic strategies aimed at the ceramide signaling pathway may offer enhanced specificity. For example, ceramide synthase inhibitors or agents that promote ceramide degradation can directly mitigate toxic lipid accumulation in the myocardium [187]. Therapies targeting inflammation pathways aim to disrupt the inflammatory responses provoked by lipotoxicity, potentially utilizing specific anti-inflammatory drugs or cytokine inhibitors to break the vicious cycle between lipotoxicity and cardiac structural remodeling [188]. Furthermore, for patients with inherited metabolic disorders or severe myocardial injury, gene therapy and cell therapy could present promising new treatment avenues.
Future research must also address several critical and challenging scientific questions:
(1) Spatiotemporal Dynamics of Lipotoxicity: How do the composition and localization of specific lipid species (e.g., ceramide subspecies, DAG) within distinct cardiomyocyte subcellular compartments (e.g., sarcolemma, mitochondrial-associated membranes, lipid droplets) dynamically change during the progression from metabolic syndrome to overt heart failure?
(2) The Lipid Droplet Paradox: What specific molecular signals govern the transition of lipid droplets from a protective storage organelle to a source of lipotoxic fatty acids? Is this due to dysregulation of the ATGL-HSL lipolytic cascade, impaired droplet autophagy (lipophagy), or alterations in the lipid droplet proteome?
(3) Personalized Metabolic Phenotyping: Can we identify specific circulating or imaging biomarkers that reliably predict an individual’s susceptibility to myocardial lipotoxicity versus their capacity for adaptive fatty acid oxidation? This would move the field towards personalized cardiometabolic medicine, allowing therapies to be tailored based on whether a patient’s heart is primarily “lipid-overloaded” or “lipid-inefficient”.
(4) Targeting Inter-organ Communication: What is the precise mechanistic role of extracellular vesicles and specific miRNAs derived from dysfunctional adipose tissue or the liver in directly instigating or amplifying lipotoxic signaling within cardiomyocytes?
(5) Rescuing Mitochondrial Quality Control: Beyond general antioxidant approaches, can we develop strategies to specifically enhance the selective autophagic clearance of lipotoxicity-damaged mitochondria (mitophagy) in the heart without disrupting overall mitochondrial biogenesis?
Large-scale prospective cohort studies, particularly those employing cardiac magnetic resonance spectroscopy (1H-MRS) technology, are essential for clarifying the quantitative relationship between myocardial lipid content and cardiovascular clinical outcomes [189]. Currently, there is a dearth of myocardium-specific biomarkers for lipotoxicity; the development of highly specific biomarkers will enhance early detection and risk stratification. Strategies to optimize the combined use of existing drugs should be refined; for instance, the combination of SGLT2 inhibitors and trimetazidine may yield synergistic effects [190]. Concurrently, the exploration of novel therapeutic targets, such as modulation of the cGAS-STING pathway or targeting specific lipotoxic lipid species, should be actively pursued [107]. It is crucial to emphasize that the formulation of any treatment strategy must be grounded in robust evidence from rigorous medical research, and the processes of clinical validation must not be overlooked or simplified.
6. Conclusions
Myocardial lipotoxicity represents a significant pathological mechanism underlying various cardiovascular diseases, particularly in the context of metabolic dysregulation. It induces cardiovascular pathologies such as diabetic cardiomyopathy, heart failure, arrhythmias, and coronary artery disease through multiple mechanisms, including excessive lipid accumulation, formation of toxic lipid species, mitochondrial dysfunction, inflammation activation, and cell death. Current assessments of this condition primarily rely on cardiac magnetic resonance spectroscopy and analysis of various biomarkers, yet highly specific detection methods remain insufficient. Therapeutic strategies encompass lifestyle modifications, the use of existing pharmacological agents (e.g., SGLT2 inhibitors and trimetazidine), and emerging targeted therapies. Future research should focus on deepening our understanding of molecular mechanisms, developing specific biomarkers, and exploring precise targeted therapeutic strategies to address the burden of cardiovascular diseases associated with myocardial lipotoxicity.
Author Contributions
Y.X. and L.F.: conceived the review; X.L.: made the figures; Y.X.: wrote the manuscript; L.F. and X.L.: reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated or analyzed in support of this review. All data discussed or cited in this manuscript are available from the original publications referenced in the reference list.
Conflicts of Interest
The authors declare no conflict of interest.
The authors declare no conflict of interest.
Use of AI and AI-Assisted Technologies: No AI tools were utilized for this paper.
References
- 1.
Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; et al. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. https://doi.org/10.1161/circresaha.121.318241.
- 2.
Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. https://doi.org/10.1161/circresaha.116.306842.
- 3.
Wenzl, F.A.; Ambrosini, S.; Mohammed, S.A.; et al. Inflammation in Metabolic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 742178. https://doi.org/10.3389/fcvm.2021.742178.
- 4.
Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 2012, 14, 517–531. https://doi.org/10.1007/s11906-012-0307-2.
- 5.
Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. https://doi.org/10.1161/circresaha.120.315913.
- 6.
Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. https://doi.org/10.1161/circresaha.118.311371.
- 7.
Hong, J.; Kim, K.; Kim, J.H.; et al. The Role of Endoplasmic Reticulum Stress in Cardiovascular Disease and Exercise. Int. J. Vasc. Med. 2017, 2017, 2049217. https://doi.org/10.1155/2017/2049217.
- 8.
Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. https://doi.org/10.1038/s41569-018-0044-6.
- 9.
Charnock, J.S. Lipids and cardiac arrhythmia. Prog. Lipid Res. 1994, 33, 355–385. https://doi.org/10.1016/0163-7827(94)90023-x.
- 10.
Libby, P.; Buring, J.E.; Badimon, L.; et al. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. https://doi.org/10.1038/s41572-019-0106-z.
- 11.
Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; et al. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fat. Acids 2010, 82, 149–154. https://doi.org/10.1016/j.plefa.2010.02.029.
- 12.
Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. https://doi.org/10.1194/jlr.R082933.
- 13.
Gianazza, E.; Brioschi, M.; Fernandez, A.M.; et al. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. https://doi.org/10.1016/j.redox.2019.101119.
- 14.
Frohnert, B.I.; Bernlohr, D.A. Regulation of fatty acid transporters in mammalian cells. Prog. Lipid Res. 2000, 39, 83–107. https://doi.org/10.1016/s0163-7827(99)00018-1.
- 15.
Chabowski, A.; Górski, J.; Glatz, J.F.; et al. Protein-mediated Fatty Acid Uptake in the Heart. Curr. Cardiol. Rev. 2008, 4, 12–21. https://doi.org/10.2174/157340308783565429.
- 16.
Shang, R.; Rodrigues, B. Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart. Biomolecules 2021, 11, 1016. https://doi.org/10.3390/biom11071016.
- 17.
Augustus, A.S.; Buchanan, J.; Park, T.S.; et al. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J. Biol. Chem. 2006, 281, 8716–8723. https://doi.org/10.1074/jbc.M509890200.
- 18.
Lee, J.; Goldberg, I.J. Lipoprotein lipase-derived fatty acids: Physiology and dysfunction. Curr. Hypertens. Rep. 2007, 9, 462–466. https://doi.org/10.1007/s11906-007-0085-4.
- 19.
Yu, J.E.; Han, S.Y.; Wolfson, B.; et al. The role of endothelial lipase in lipid metabolism, inflammation, and cancer. Histol. Histopathol. 2018, 33, 1–10. https://doi.org/10.14670/hh-11-905.
- 20.
Yasuda, T.; Ishida, T.; Rader, D.J. Update on the role of endothelial lipase in high-density lipoprotein metabolism, reverse cholesterol transport, and atherosclerosis. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 2263–2270. https://doi.org/10.1253/circj.cj-10-0934.
- 21.
Nakajima, H.; Ishida, T.; Satomi-Kobayashi, S.; et al. Endothelial lipase modulates pressure overload-induced heart failure through alternative pathway for fatty acid uptake. Hypertension 2013, 61, 1002–1007. https://doi.org/10.1161/hypertensionaha.111.201608.
- 22.
Fujimoto, T.; Parton, R.G. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol. 2011, 3, a004838. https://doi.org/10.1101/cshperspect.a004838.
- 23.
Steinbusch, L.K.; Wijnen, W.; Schwenk, R.W.; et al. Differential regulation of cardiac glucose and fatty acid uptake by endosomal pH and actin filaments. Am. J. Physiol. Cell Physiol. 2010, 298, C1549–C1559. https://doi.org/10.1152/ajpcell.00334.2009.
- 24.
Ouwens, D.M.; Diamant, M.; Fodor, M.; et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007, 50, 1938–1948. https://doi.org/10.1007/s00125-007-0735-8.
- 25.
Zorzano, A.; Fandos, C.; Palacín, M. Role of plasma membrane transporters in muscle metabolism. Biochem. J. 2000, 349, 667–688. https://doi.org/10.1042/bj3490667.
- 26.
Stremmel, W. Fatty acid uptake by isolated rat heart myocytes represents a carrier-mediated transport process. J. Clin. Investig. 1988, 81, 844–852. https://doi.org/10.1172/jci113393.
- 27.
Luiken, J.J.; van Nieuwenhoven, F.A.; America, G.; et al. Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: Involvement of sarcolemmal proteins. J. Lipid Res. 1997, 38, 745–758.
- 28.
Jain, S.S.; Chabowski, A.; Snook, L.A.; et al. Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FABPpm, FATP1, 4 and 6. FEBS Lett. 2009, 583, 2294–2300. https://doi.org/10.1016/j.febslet.2009.06.020.
- 29.
Hajri, T.; Abumrad, N.A. Fatty acid transport across membranes: Relevance to nutrition and metabolic pathology. Annu. Rev. Nutr. 2002, 22, 383–415. https://doi.org/10.1146/annurev.nutr.22.020402.130846.
- 30.
Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. https://doi.org/10.1016/j.cmet.2012.04.006.
- 31.
Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. https://doi.org/10.1038/nrd2589.
- 32.
Palomer, X.; Salvadó, L.; Barroso, E.; et al. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. https://doi.org/10.1016/j.ijcard.2013.07.150.
- 33.
McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—Old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. https://doi.org/10.1038/nrendo.2015.129.
- 34.
Ellis, J.M.; Frahm, J.L.; Li, L.O.; et al. Acyl-coenzyme A synthetases in metabolic control. Curr. Opin. Lipidol. 2010, 21, 212–217. https://doi.org/10.1097/mol.0b013e32833884bb.
- 35.
Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. https://doi.org/10.1007/s10545-010-9061-2.
- 36.
Da Dalt, L.; Cabodevilla, A.G.; Goldberg, I.J.; et al. Cardiac lipid metabolism, mitochondrial function, and heart failure. Cardiovasc. Res. 2023, 119, 1905–1914. https://doi.org/10.1093/cvr/cvad100.
- 37.
Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. https://doi.org/10.1016/j.bbalip.2011.09.010.
- 38.
Glatz, J.F.; Nabben, M.; Heather, L.C.; et al. Regulation of the subcellular trafficking of CD36, a major determinant of cardiac fatty acid utilization. Biochim. Biophys. Acta 2016, 1861, 1461–1471. https://doi.org/10.1016/j.bbalip.2016.04.008.
- 39.
Luiken, J.J.; Coort, S.L.; Willems, J.; et al. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 2003, 52, 1627–1634. https://doi.org/10.2337/diabetes.52.7.1627.
- 40.
Steinbusch, L.K.; Schwenk, R.W.; Ouwens, D.M.; et al. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell. Mol. Life Sci. CMLS 2011, 68, 2525–2538. https://doi.org/10.1007/s00018-011-0690-x.
- 41.
Tong, F.; Black, P.N.; Coleman, R.A.; et al. Fatty acid transport by vectorial acylation in mammals: Roles played by different isoforms of rat long-chain acyl-CoA synthetases. Arch. Biochem. Biophys. 2006, 447, 46–52. https://doi.org/10.1016/j.abb.2006.01.005.
- 42.
Zhang, Q.; Li, J.; Liu, X.; et al. Inhibiting CD36 palmitoylation improves cardiac function post-infarction by regulating lipid metabolic homeostasis and autophagy. Nat. Commun. 2025, 16, 6602. https://doi.org/10.1038/s41467-025-61875-y.
- 43.
Geng, J.; Zhang, X.; Wang, Y.; et al. CD36 knockdown attenuates pressure overload-induced cardiac injury by preventing lipotoxicity and improving myocardial energy metabolism. Int. J. Med. Sci. 2025, 22, 1223–1236. https://doi.org/10.7150/ijms.107224.
- 44.
Chiu, H.C.; Kovacs, A.; Blanton, R.M.; et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 2005, 96, 225–233. https://doi.org/10.1161/01.Res.0000154079.20681.B9.
- 45.
Thompson, B.R.; Lobo, S.; Bernlohr, D.A. Fatty acid flux in adipocytes: The in's and out's of fat cell lipid trafficking. Mol. Cell. Endocrinol. 2010, 318, 24–33. https://doi.org/10.1016/j.mce.2009.08.015.
- 46.
Black, P.N.; Sandoval, A.; Arias-Barrau, E.; et al. Targeting the fatty acid transport proteins (FATP) to understand the mechanisms linking fatty acid transport to metabolism. Immunol. Endocr. Metab. Agents Med. Chem. 2009, 9, 11–17. https://doi.org/10.2174/187152209788009850.
- 47.
Hendgen-Cotta, U.B.; Esfeld, S.; Coman, C.; et al. A novel physiological role for cardiac myoglobin in lipid metabolism. Sci. Rep. 2017, 7, 43219. https://doi.org/10.1038/srep43219.
- 48.
Pei, Z.; Deng, Q.; Babcock, S.A.; et al. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: Role of autophagy and ER stress. Toxicol. Lett. 2018, 284, 10–20. https://doi.org/10.1016/j.toxlet.2017.11.018.
- 49.
Furuhashi, M. Fatty Acid-Binding Protein 4 in Cardiovascular and Metabolic Diseases. J. Atheroscler. Thromb. 2019, 26, 216–232. https://doi.org/10.5551/jat.48710.
- 50.
Bertinchant, J.P.; Polge, A. Diagnostic and prognostic value of heart-type fatty acid-binding protein (H-FABP), an early biochemical marker of myocardial injury. Arch. Mal. Coeur Vaiss. 2005, 98, 1225–1231.
- 51.
Ellis, J.M.; Li, L.O.; Wu, P.C.; et al. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab. 2010, 12, 53–64. https://doi.org/10.1016/j.cmet.2010.05.012.
- 52.
Durgan, D.J.; Smith, J.K.; Hotze, M.A.; et al. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2480–H2497. https://doi.org/10.1152/ajpheart.01344.2005.
- 53.
Zhao, Z.D.; Zan, L.S.; Li, A.N.; et al. Characterization of the promoter region of the bovine long-chain acyl-CoA synthetase 1 gene: Roles of E2F1, Sp1, KLF15, and E2F4. Sci. Rep. 2016, 6, 19661. https://doi.org/10.1038/srep19661.
- 54.
Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. https://doi.org/10.1210/endocr/bqz046.
- 55.
Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. https://doi.org/10.1038/s41580-018-0085-z.
- 56.
Trites, M.J.; Clugston, R.D. The role of adipose triglyceride lipase in lipid and glucose homeostasis: Lessons from transgenic mice. Lipids Health Dis. 2019, 18, 204. https://doi.org/10.1186/s12944-019-1151-z.
- 57.
Lass, A.; Zimmermann, R.; Oberer, M.; et al. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. https://doi.org/10.1016/j.plipres.2010.10.004.
- 58.
Grabner, G.F.; Xie, H.; Schweiger, M.; et al. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. https://doi.org/10.1038/s42255-021-00493-6.
- 59.
Kartsoli, S.; Kostara, C.E.; Tsimihodimos, V.; et al. Lipidomics in non-alcoholic fatty liver disease. World J. Hepatol. 2020, 12, 436–450. https://doi.org/10.4254/wjh.v12.i8.436.
- 60.
Pei, K.; Gui, T.; Kan, D.; et al. An Overview of Lipid Metabolism and Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2020, 2020, 4020249. https://doi.org/10.1155/2020/4020249.
- 61.
Woodcock, J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life 2006, 58, 462–466. https://doi.org/10.1080/15216540600871118.
- 62.
Bekhite, M.; González-Delgado, A.; Hübner, S.; et al. The role of ceramide accumulation in human induced pluripotent stem cell-derived cardiomyocytes on mitochondrial oxidative stress and mitophagy. Free Radic. Biol. Med. 2021, 167, 66–80. https://doi.org/10.1016/j.freeradbiomed.2021.02.016.
- 63.
Liu, W.; Ruiz-Velasco, A.; Wang, S.; et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat. Commun. 2017, 8, 494. https://doi.org/10.1038/s41467-017-00664-8.
- 64.
Park, T.S.; Hu, Y.; Noh, H.L.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. https://doi.org/10.1194/jlr.M800147-JLR200.
- 65.
Basu, R.; Oudit, G.Y.; Wang, X.; et al. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2096–H2108. https://doi.org/10.1152/ajpheart.00452.2009.
- 66.
Carrasco, S.; Mérida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. https://doi.org/10.1016/j.tibs.2006.11.004.
- 67.
Riehle, C.; Abel, E.D. Insulin Signaling and Heart Failure. Circ. Res. 2016, 118, 1151–1169. https://doi.org/10.1161/circresaha.116.306206.
- 68.
Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; et al. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. https://doi.org/10.1111/febs.13709.
- 69.
Battiprolu, P.K.; Hojayev, B.; Jiang, N.; et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Investig. 2012, 122, 1109–1118. https://doi.org/10.1172/jci60329.
- 70.
Singh, R.M.; Cummings, E.; Pantos, C.; et al. Protein kinase C and cardiac dysfunction: A review. Heart Fail. Rev. 2017, 22, 843–859. https://doi.org/10.1007/s10741-017-9634-3.
- 71.
Sakane, F.; Mizuno, S.; Komenoi, S. Diacylglycerol Kinases as Emerging Potential Drug Targets for a Variety of Diseases: An Update. Front. Cell Dev. Biol. 2016, 4, 82. https://doi.org/10.3389/fcell.2016.00082.
- 72.
Shreya, S.; Alam, M.J.; Anupriya; et al. Lipotoxicity, ER Stress, and Cardiovascular Disease: Current Understanding and Future Directions. Cardiovasc. Hematol. Agents Med. Chem. 2024, 22, 319–335. https://doi.org/10.2174/0118715257262366230928051902.
- 73.
Tokuyama, T.; Yanagi, S. Role of Mitochondrial Dynamics in Heart Diseases. Genes 2023, 14, 1876. https://doi.org/10.3390/genes14101876.
- 74.
Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. https://doi.org/10.1016/j.jacc.2012.08.1021.
- 75.
Busiello, R.A.; Savarese, S.; Lombardi, A. Mitochondrial uncoupling proteins and energy metabolism. Front. Physiol. 2015, 6, 36. https://doi.org/10.3389/fphys.2015.00036.
- 76.
Erlanson-Albertsson, C. The role of uncoupling proteins in the regulation of metabolism. Acta Physiol. Scand. 2003, 178, 405–412. https://doi.org/10.1046/j.1365-201X.2003.01159.x.
- 77.
Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. https://doi.org/10.1016/j.tem.2015.12.001.
- 78.
Smirnova, E.; Griparic, L.; Shurland, D.L.; et al. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. https://doi.org/10.1091/mbc.12.8.2245.
- 79.
Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; et al. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998, 143, 351–358. https://doi.org/10.1083/jcb.143.2.351.
- 80.
Ong, S.B.; Subrayan, S.; Lim, S.Y.; et al. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. https://doi.org/10.1161/circulationaha.109.906610.
- 81.
Bean, C.; Audano, M.; Varanita, T.; et al. The mitochondrial protein Opa1 promotes adipocyte browning that is dependent on urea cycle metabolites. Nat. Metab. 2021, 3, 1633–1647. https://doi.org/10.1038/s42255-021-00497-2.
- 82.
Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. CMLS 2016, 73, 79–94. https://doi.org/10.1007/s00018-015-2052-6.
- 83.
Wang, S.; Binder, P.; Fang, Q.; et al. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. https://doi.org/10.1111/bph.13888.
- 84.
Ozcan, L.; Tabas, I. Calcium signalling and ER stress in insulin resistance and atherosclerosis. J. Intern. Med. 2016, 280, 457–464. https://doi.org/10.1111/joim.12562.
- 85.
Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. https://doi.org/10.1038/s41580-020-0250-z.
- 86.
Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. https://doi.org/10.1126/science.aat5314.
- 87.
B'Chir, W.; Maurin, A.C.; Carraro, V.; et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. https://doi.org/10.1093/nar/gkt563.
- 88.
Gong, J.; Wang, L.; Tao, W.; et al. AMPK Mediates Early Activation of the Unfolded Protein Response through a Positive Feedback Loop in Palmitate-Treated Muscle Cells. Front. Biosci. 2023, 28, 159. https://doi.org/10.31083/j.fbl2808159.
- 89.
Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. https://doi.org/10.1016/s0092-8674(00)80369-4.
- 90.
Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039.
- 91.
Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. https://doi.org/10.1016/j.molcel.2017.06.017.
- 92.
Congur, I.; Mingrone, G.; Guan, K. Targeting endoplasmic reticulum stress as a potential therapeutic strategy for diabetic cardiomyopathy. Metab. Clin. Exp. 2025, 162, 156062. https://doi.org/10.1016/j.metabol.2024.156062.
- 93.
Turpin, J.; El-Safadi, D.; Lebeau, G.; et al. CHOP Pro-Apoptotic Transcriptional Program in Response to ER Stress Is Hacked by Zika Virus. Int. J. Mol. Sci. 2021, 22, 3750. https://doi.org/10.3390/ijms22073750.
- 94.
Wen, H.; Gris, D.; Lei, Y.; et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. https://doi.org/10.1038/ni.2022.
- 95.
Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. https://doi.org/10.1155/2014/360438.
- 96.
Mih, N.; Monk, J.M.; Fang, X.; et al. Adaptations of Escherichia coli strains to oxidative stress are reflected in properties of their structural proteomes. BMC Bioinform. 2020, 21, 162. https://doi.org/10.1186/s12859-020-3505-y.
- 97.
Zhang, T.; Zhang, Y.; Cui, M.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. https://doi.org/10.1038/nm.4017.
- 98.
Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. https://doi.org/10.1038/nrcardio.2017.161.
- 99.
Chiu, H.C.; Kovacs, A.; Ford, D.A.; et al. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. https://doi.org/10.1172/jci10947.
- 100.
Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. https://doi.org/10.1172/jci88883.
- 101.
Young, S.G.; Fong, L.G.; Beigneux, A.P.; et al. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 2019, 30, 51–65. https://doi.org/10.1016/j.cmet.2019.05.023.
- 102.
Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. https://doi.org/10.1152/physrev.00006.2004.
- 103.
Sun, Q.; Karwi, Q.G.; Wong, N.; et al. Advances in myocardial energy metabolism: Metabolic remodelling in heart failure and beyond. Cardiovasc. Res. 2024, 120, 1996–2016. https://doi.org/10.1093/cvr/cvae231.
- 104.
Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. https://doi.org/10.1007/s11897-013-0133-0.
- 105.
Bi, X.; Wu, X.; Chen, J.; et al. Characterization of ferroptosis-triggered pyroptotic signaling in heart failure. Signal Transduct. Target. Ther. 2024, 9, 257. https://doi.org/10.1038/s41392-024-01962-6.
- 106.
Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; et al. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. https://doi.org/10.1152/physrev.00015.2009.
- 107.
Yan, M.; Li, Y.; Luo, Q.; et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022, 8, 258. https://doi.org/10.1038/s41420-022-01046-w.
- 108.
Chumakova, G.; Gritsenko, O.; Gruzdeva, O.; et al. Analysis of probable lipotoxic damage and myocardial fibrosis in epicardial obesity. Aging 2021, 13, 14806–14815. https://doi.org/10.18632/aging.203148.
- 109.
Zhao, M.; Bian, R.; Xu, X.; et al. Sphingolipid Metabolism and Signalling Pathways in Heart Failure: From Molecular Mechanism to Therapeutic Potential. J. Inflamm. Res. 2025, 18, 5477–5498. https://doi.org/10.2147/jir.S515757.
- 110.
Law, B.A.; Liao, X.; Moore, K.S.; et al. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 1403–1416. https://doi.org/10.1096/fj.201700300R.
- 111.
Ramírez-Camacho, I.; Bautista-Pérez, R.; Correa, F.; et al. Role of sphingomyelinase in mitochondrial ceramide accumulation during reperfusion. Biochim. Biophys. Acta 2016, 1862, 1955–1963. https://doi.org/10.1016/j.bbadis.2016.07.021.
- 112.
Park, T.S.; Goldberg, I.J. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail. Clin. 2012, 8, 633–641. https://doi.org/10.1016/j.hfc.2012.06.003.
- 113.
Chokshi, A.; Drosatos, K.; Cheema, F.H.; et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012, 125, 2844–2853. https://doi.org/10.1161/circulationaha.111.060889.
- 114.
Zhuang, L.; Li, C.; Chen, Q.; et al. Fatty acid-binding protein 3 contributes to ischemic heart injury by regulating cardiac myocyte apoptosis and MAPK pathways. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H971-H984. https://doi.org/10.1152/ajpheart.00360.2018.
- 115.
D'Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim. Biophys. Acta 2016, 1861, 1513–1524. https://doi.org/10.1016/j.bbalip.2016.02.016.
- 116.
Matsui, T.; Tao, J.; del Monte, F.; et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 2001, 104, 330–335. https://doi.org/10.1161/01.cir.104.3.330.
- 117.
Steinberg, S.F. Cardiac actions of protein kinase C isoforms. Physiology 2012, 27, 130–139. https://doi.org/10.1152/physiol.00009.2012.
- 118.
Paz, K.; Liu, Y.F.; Shorer, H.; et al. Phosphorylation of insulin receptor substrate-1 (IRS-1) by protein kinase B positively regulates IRS-1 function. J. Biol. Chem. 1999, 274, 28816–28822. https://doi.org/10.1074/jbc.274.40.28816.
- 119.
Kim, J.K.; Fillmore, J.J.; Sunshine, M.J.; et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J. Clin. Investig. 2004, 114, 823–827. https://doi.org/10.1172/jci22230.
- 120.
Marx, S.O.; Marks, A.R. Regulation of the ryanodine receptor in heart failure. Basic Res. Cardiol. 2002, 97 (Suppl. 1), I49–I51. https://doi.org/10.1007/s003950200029.
- 121.
Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. https://doi.org/10.1161/circresaha.111.259754.
- 122.
Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J. Intern. Med. 2018, 284, 478–491. https://doi.org/10.1111/joim.12728.
- 123.
Kok, B.P.; Brindley, D.N. Myocardial fatty acid metabolism and lipotoxicity in the setting of insulin resistance. Heart Fail. Clin. 2012, 8, 643–661. https://doi.org/10.1016/j.hfc.2012.06.008.
- 124.
Cheng, L.; Ding, G.; Qin, Q.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. https://doi.org/10.1038/nm1116.
- 125.
Russell, R.R., 3rd; Li, J.; Coven, D.L.; et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. https://doi.org/10.1172/jci19297.
- 126.
Li, B.; Rao, J.; Hu, W.; et al. Adropin as a therapeutic candidate for HFpEF: Evidence of oxidative stress mitigation via Nrf2/HO-1 signaling. Lipids Health Dis. 2025, 24, 273. https://doi.org/10.1186/s12944-025-02703-6.
- 127.
Lei, M.; Salvage, S.C.; Jackson, A.P.; et al. Cardiac arrhythmogenesis: Roles of ion channels and their functional modification. Front. Physiol. 2024, 15, 1342761. https://doi.org/10.3389/fphys.2024.1342761.
- 128.
Zietzer, A.; Düsing, P.; Reese, L.; et al. Ceramide Metabolism in Cardiovascular Disease: A Network With High Therapeutic Potential. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1220–1228. https://doi.org/10.1161/atvbaha.122.318048.
- 129.
Wickenden, A.D.; Kaprielian, R.; Kassiri, Z.; et al. The role of action potential prolongation and altered intracellular calcium handling in the pathogenesis of heart failure. Cardiovasc. Res. 1998, 37, 312–323. https://doi.org/10.1016/s0008-6363(97)00256-3.
- 130.
Gowen, B.H.; Reyes, M.V.; Joseph, L.C.; et al. Mechanisms of Chronic Metabolic Stress in Arrhythmias. Antioxidants 2020, 9, 1012. https://doi.org/10.3390/antiox9101012.
- 131.
Wehrens, X.H. CaMKII regulation of the cardiac ryanodine receptor and sarcoplasmic reticulum calcium release. Heart Rhythm 2011, 8, 323–325. https://doi.org/10.1016/j.hrthm.2010.09.079.
- 132.
De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; et al. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. https://doi.org/10.3390/antiox12020353.
- 133.
Grimsrud, P.A.; Xie, H.; Griffin, T.J.; et al. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. https://doi.org/10.1074/jbc.R700019200.
- 134.
Bellinger, A.M.; Reiken, S.; Dura, M.; et al. Remodeling of ryanodine receptor complex causes “leaky” channels: A molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 2198–2202. https://doi.org/10.1073/pnas.0711074105.
- 135.
Bers, D.M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014, 76, 107–127. https://doi.org/10.1146/annurev-physiol-020911-153308.
- 136.
Slawik, M.; Vidal-Puig, A.J. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res. Rev. 2006, 5, 144–164. https://doi.org/10.1016/j.arr.2006.03.004.
- 137.
Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; et al. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. https://doi.org/10.1161/circresaha.117.310230.
- 138.
Weiss, J.N.; Nivala, M.; Garfinkel, A.; et al. Alternans and arrhythmias: From cell to heart. Circ. Res. 2011, 108, 98–112. https://doi.org/10.1161/circresaha.110.223586.
- 139.
Lewek, J.; Kaczmarek, K.; Cygankiewicz, I.; et al. Inflammation and arrhythmias: Potential mechanisms and clinical implications. Expert Rev. Cardiovasc. Ther. 2014, 12, 1077–1085. https://doi.org/10.1586/14779072.2014.942286.
- 140.
Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. https://doi.org/10.1161/01.res.87.8.656.
- 141.
Shu, H.; Cheng, J.; Li, N.; et al. Obesity and atrial fibrillation: A narrative review from arrhythmogenic mechanisms to clinical significance. Cardiovasc. Diabetol. 2023, 22, 192. https://doi.org/10.1186/s12933-023-01913-5.
- 142.
Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. https://doi.org/10.1038/s41573-021-00198-1.
- 143.
Borén, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. https://doi.org/10.1097/mol.0000000000000330.
- 144.
Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. https://doi.org/10.1155/2013/152786.
- 145.
Jiang, H.; Zhou, Y.; Nabavi, S.M.; et al. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. https://doi.org/10.3389/fcvm.2022.925923.
- 146.
Chen, C.; Khismatullin, D.B. Oxidized low-density lipoprotein contributes to atherogenesis via co-activation of macrophages and mast cells. PLoS ONE 2015, 10, e0123088. https://doi.org/10.1371/journal.pone.0123088.
- 147.
Navarro, E.; Mallén, A.; Cruzado, J.M.; et al. Unveiling ncRNA regulatory axes in atherosclerosis progression. Clin. Transl. Med. 2020, 9, 5. https://doi.org/10.1186/s40169-020-0256-3.
- 148.
Sheikh, M.S.A. Role of Plasma Soluble Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 and microRNA-98 in Severity and Risk of Coronary Artery Disease. Balk. Med. J. 2021, 38, 13–22. https://doi.org/10.5152/balkanmedj.2021.20243.
- 149.
Banerjee, D.; Sinha, A.; Saikia, S.; et al. Inflammation-induced mTORC2-Akt-mTORC1 signaling promotes macrophage foam cell formation. Biochimie 2018, 151, 139–149. https://doi.org/10.1016/j.biochi.2018.06.001.
- 150.
Michael, D.R.; Davies, T.S.; Laubertová, L.; et al. The phosphoinositide 3-kinase signaling pathway is involved in the control of modified low-density lipoprotein uptake by human macrophages. Lipids 2015, 50, 253–260. https://doi.org/10.1007/s11745-015-3993-0.
- 151.
Miller, Y.I.; Choi, S.H.; Fang, L.; et al. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc. Med. 2009, 19, 227–232. https://doi.org/10.1016/j.tcm.2010.02.001.
- 152.
Xu, X.H.; Shah, P.K.; Faure, E.; et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104, 3103–3108. https://doi.org/10.1161/hc5001.100631.
- 153.
Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. https://doi.org/10.1097/00041433-200306000-00008.
- 154.
McLaren, J.E.; Michael, D.R.; Ashlin, T.G.; et al. Cytokines, macrophage lipid metabolism and foam cells: Implications for cardiovascular disease therapy. Prog. Lipid Res. 2011, 50, 331–347. https://doi.org/10.1016/j.plipres.2011.04.002.
- 155.
Singh, R.K.; Haka, A.S.; Asmal, A.; et al. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 86–102. https://doi.org/10.1161/atvbaha.119.313200.
- 156.
Steinberg, G.R.; Schertzer, J.D. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: Implications for diabetes and cardiovascular disease. Immunol. Cell Biol. 2014, 92, 340–345. https://doi.org/10.1038/icb.2014.11.
- 157.
Li, B.R.; Xia, L.Q.; Liu, J.; et al. miR-758-5p regulates cholesterol uptake via targeting the CD36 3'UTR. Biochem. Biophys. Res. Commun. 2017, 494, 384–389. https://doi.org/10.1016/j.bbrc.2017.09.150.
- 158.
Yalcinkaya, M.; Tall, A.R. Cholesterol Crystals as Triggers of NLRP3 Inflammasome Activation in Atherosclerosis. Curr. Atheroscler. Rep. 2025, 27, 77. https://doi.org/10.1007/s11883-025-01323-w.
- 159.
Su, S.H.; Wu, Y.F.; Lin, Q.; et al. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflamm. 2019, 16, 260. https://doi.org/10.1186/s12974-019-1668-0.
- 160.
Yu, Y.Y.; Wang, R.; Chen, G.Q.; et al. Mechanisms and Targeted Therapeutic Strategies in Sepsis-Induced Myocardial Dysfunction: The Role of NLRP3 Inflammasome-Mediated Inflammation. J. Inflamm. Res. 2025, 18, 8875–8897. https://doi.org/10.2147/jir.S521655.
- 161.
Hu, Q.; Zhang, T.; Yi, L.; et al. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. BioFactors 2018, 44, 123–136. https://doi.org/10.1002/biof.1395.
- 162.
Hoseini, Z.; Sepahvand, F.; Rashidi, B.; et al. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. https://doi.org/10.1002/jcp.25930.
- 163.
Rajamäki, K.; Lappalainen, J.; Oörni, K.; et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. https://doi.org/10.1371/journal.pone.0011765.
- 164.
Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. https://doi.org/10.1161/atvbaha.107.159327.
- 165.
Shimokado, K.; Raines, E.W.; Madtes, D.K.; et al. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell 1985, 43, 277–286. https://doi.org/10.1016/0092-8674(85)90033-9.
- 166.
Daoud, F.; Arévalo Martínez, M.; Holst, J.; et al. Role of smooth muscle YAP and TAZ in protection against phenotypic modulation, inflammation, and aneurysm development. Biochem. Pharmacol. 2022, 206, 115307. https://doi.org/10.1016/j.bcp.2022.115307.
- 167.
Jin, P.; Duan, X.; Huang, Z.; et al. Nuclear receptors in health and disease: Signaling pathways, biological functions and pharmaceutical interventions. Signal Transduct. Target. Ther. 2025, 10, 228. https://doi.org/10.1038/s41392-025-02270-3.
- 168.
Zhang, Y.; Zeng, M.; Zhang, X.; et al. Tiaogan daozhuo formula attenuates atherosclerosis via activating AMPK -PPARγ-LXRα pathway. J. Ethnopharmacol. 2024, 324, 117814. https://doi.org/10.1016/j.jep.2024.117814.
- 169.
Li, Y.; Zhang, L.; Ren, P.; et al. Qing-Xue-Xiao-Zhi formula attenuates atherosclerosis by inhibiting macrophage lipid accumulation and inflammatory response via TLR4/MyD88/NF-κB pathway regulation. Phytomedicine Int. J. Phytother. Phytopharm. 2021, 93, 153812. https://doi.org/10.1016/j.phymed.2021.153812.
- 170.
Qian, T.; Guo, D.; Sun, L.; et al. Crosstalk between lipid metabolism and macrophages in atherosclerosis: Therapeutic potential of natural products. Front. Cardiovasc. Med. 2025, 12, 1529924. https://doi.org/10.3389/fcvm.2025.1529924.
- 171.
Zhang, H.; Sáenz de Urturi, D.; Fernández-Tussy, P.; et al. Hypercholesterolemia-induced LXR signaling in smooth muscle cells contributes to vascular lesion remodeling and visceral function. Proc. Natl. Acad. Sci. USA 2025, 122, e2417512122. https://doi.org/10.1073/pnas.2417512122.
- 172.
Bechmann, L.E.; Emanuelsson, F.; Nordestgaard, B.G.; et al. SGLT2-inhibition increases total, LDL, and HDL cholesterol and lowers triglycerides: Meta-analyses of 60 randomized trials, overall and by dose, ethnicity, and drug type. Atherosclerosis 2024, 394, 117236. https://doi.org/10.1016/j.atherosclerosis.2023.117236.
- 173.
McGuire, D.K.; Shih, W.J.; Cosentino, F.; et al. Association of SGLT2 Inhibitors With Cardiovascular and Kidney Outcomes in Patients with Type 2 Diabetes: A Meta-analysis. JAMA Cardiol. 2021, 6, 148–158. https://doi.org/10.1001/jamacardio.2020.4511.
- 174.
Delanaye, P.; Scheen, A.J. The diuretic effects of SGLT2 inhibitors: A comprehensive review of their specificities and their role in renal protection. Diabetes Metab. 2021, 47, 101285. https://doi.org/10.1016/j.diabet.2021.101285.
- 175.
Marzilli, M.; Klein, W.W. Efficacy and tolerability of trimetazidine in stable angina: A meta-analysis of randomized, double-blind, controlled trials. Coron. Artery Dis. 2003, 14, 171–179. https://doi.org/10.1097/00019501-200304000-00010.
- 176.
Chazov, E.I.; Lepakchin, V.K.; Zharova, E.A.; et al. Trimetazidine in Angina Combination Therapy-the TACT study: Trimetazidine versus conventional treatment in patients with stable angina pectoris in a randomized, placebo-controlled, multicenter study. Am. J. Ther. 2005, 12, 35–42. https://doi.org/10.1097/00045391-200501000-00006.
- 177.
Dyck, J.R.B.; Sossalla, S.; Hamdani, N.; et al. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: Evidence for potential off-target effects. J. Mol. Cell. Cardiol. 2022, 167, 17–31. https://doi.org/10.1016/j.yjmcc.2022.03.005.
- 178.
Rosano, G.M.; Vitale, C. Metabolic Modulation of Cardiac Metabolism in Heart Failure. Card. Fail. Rev. 2018, 4, 99–103. https://doi.org/10.15420/cfr.2018.18.2.
- 179.
Goel, H.; Roma, N.; Morgan, M.; et al. Trimetazidine in Cardiovascular Disease and Beyond: A Comprehensive Review. Am. J. Cardiovasc. Drugs Drugs 2025, 25, 443–460. https://doi.org/10.1007/s40256-025-00724-1.
- 180.
Staels, B.; Dallongeville, J.; Auwerx, J.; et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. https://doi.org/10.1161/01.cir.98.19.2088.
- 181.
Braissant, O.; Foufelle, F.; Scotto, C.; et al. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. https://doi.org/10.1210/endo.137.1.8536636.
- 182.
Fruchart, J.C. Pemafibrate (K-877), a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc. Diabetol. 2017, 16, 124. https://doi.org/10.1186/s12933-017-0602-y.
- 183.
Das Pradhan, A.; Glynn, R.J.; Fruchart, J.C.; et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. New Engl. J. Med. 2022, 387, 1923–1934. https://doi.org/10.1056/NEJMoa2210645.
- 184.
Morris, R.; Bu, K.; Han, W.; et al. The Association Between Statin Drugs and Rhabdomyolysis: An Analysis of FDA Adverse Event Reporting System (FAERS) Data and Transcriptomic Profiles. Genes 2025, 16, 248. https://doi.org/10.3390/genes16030248.
- 185.
Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. https://doi.org/10.1152/physrev.00063.2017.
- 186.
Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; et al. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. https://doi.org/10.1126/science.1056843.
- 187.
Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. https://doi.org/10.1210/er.2007-0025.
- 188.
Łoboda, A.; Dulak, J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: Past, present, and future. Pharmacol. Rep. 2020, 72, 1227–1263. https://doi.org/10.1007/s43440-020-00134-x.
- 189.
Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.J.; et al. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 2003, 49, 417–423. https://doi.org/10.1002/mrm.10372.
- 190.
Honka, H.; Solis-Herrera, C.; Triplitt, C.; et al. Therapeutic Manipulation of Myocardial Metabolism: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 2022–2039. https://doi.org/10.1016/j.jacc.2021.02.057.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com