The nucleolus is recognized as the largest and most architecturally complex membrane-less organelle within the mammalian nucleus, exhibiting pronounced structural dynamics. Notably, this compartment demonstrates exceptional sensitivity to cellular stress; such perturbations frequently culminate in nucleolar stress, a condition characterized by structural disintegration, functional compromise, and organellar destabilization. Nucleolar stress has emerged as a critical paradigm, positing the nucleolus as both a stress sensor and a signaling hub under pathological conditions. Mechanistically, nucleolar stress responses have been demonstrated to exert pleiotropic regulatory effects on cell cycle progression, differentiation and cell fate determination, thereby triggering apoptosis, senescence, or autophagy in stressed cells. The nucleolus, being the principal site of ribosomal biogenesis and cell cycle control, has been implicated in the pathogenesis of cardiovascular disorders. Clinical and experimental evidence consistently reveals distinct nucleolar morphological aberrations and ribosomal dysfunction during cardiovascular stress events, particularly in myocardial infarction and cardiomyopathy. These disruptions have been shown to impair cardiac proteostasis and metabolic homeostasis, consequently exacerbating myocardial dysfunction. Therefore, elucidating the molecular mechanisms underlying stress-induced nucleolar signaling pathways may provide two key translational benefits: the identification of novel diagnostic biomarkers for early cardiovascular disease detection, and the discovery of precision therapeutic targets. Such advancements could substantially refine clinical management strategies and improve patients’ prognoses.
- Open Access
- Review
Nucleolus, Nucleolar Stress and Cardiovascular Diseases
- Rongqian Xu 1,
- Xiaocen Jin 1,
- Yan Zhang 2,
- Xiaolei Yang 1,
- Yunlong Xia 1,*
Author Information
Received: 10 Jun 2025 | Revised: 07 Aug 2025 | Accepted: 15 Aug 2025 | Published: 29 Jan 2026
Abstract
Keywords
nucleolus | nucleolar proteins | cardiovascular diseases | stress events | liquid-liquid phase separation
1. Introduction
The nucleolus, a prominent membraneless organelle within the eukaryotic nucleus, serves as the primary site for ribosomal biogenesis while playing pivotal roles in diverse cellular processes, including cell cycle control and stress sensing. Emerging evidence has revealed that nucleolar stress—a pathophysiological state triggered by structural or functional perturbations of the nucleolus—exerts profound influences on the pathogenesis and progression of cardiovascular diseases (CVDs). Through multifaceted mechanisms, nucleolar stress disrupts cardiovascular homeostasis, contributing to disease development. Current therapeutic strategies are increasingly targeting nucleolar stress-associated signaling cascades, particularly the well-studied p53 and mTOR pathways, to mitigate cardiovascular injury. Nevertheless, the precise molecular mechanisms governing nucleolar involvement in CVDs and their potential clinical translation remain incompletely understood. Functioning as a critical nexus between fundamental cellular biology and cardiovascular pathophysiology, nucleolar stress offers novel insights into disease mechanisms and therapeutic opportunities. A deeper mechanistic understanding of nucleolar stress may pave the way for identifying innovative diagnostic biomarkers and targeted interventions, ultimately improving clinical outcomes in CVD patients. This review systematically illustrates the structural and functional features of the nucleolus, delineates the hallmarks of nucleolar stress responses, and elucidates their mechanistic links to cardiovascular diseases.
2. Nucleolus Characteristics
The nucleolus was first identified in the 1830s by physiologist Gabriel Gustav Valentin, who observed a highly refractive intranuclear body using light microscopy. Notably, due to its central nuclear localization, this structure was designated the “nucleolus” [1]. Subsequent investigations in the 1930s by Heitz and McClintock demonstrated that nucleolar assembly occurs at specific chromosomal loci [2,3], later characterized as nucleolar organizer regions (NORs) [4]. These domains were subsequently shown to comprise clustered ribosomal deoxyribonucleic acid (rDNA) transcription units [5], which are invariably surrounded by perinucleolar heterochromatin [6]. From a structural perspective, the nucleolus is recognized as the most prominent and evolutionarily conserved membrane-less subnuclear compartment in eukaryotes [7]. Ultrastructural analyses via electron microscopy have resolved its tripartite architecture, consisting of three concentric subdomains: (1) the fibrillar center (FC), the site of rDNA transcription, (2) the dense fibrillar component (DFC), where ribosomal ribonucleic acid (rRNA) processing occur, and (3) the granular component (GC), which facilitates ribosome assembly [5]. However, the spatial organization of these compartments exhibits considerable heterogeneity across species, cell types, and physiological conditions [8]. For instance, Scheer et al. reported striking organizational differences: nucleoli in mouse Ehrlich ascites tumor cells display a tightly packed concentric arrangement, whereas those in cultured RV rat cells adopt a reticular configuration featuring multiple small FCs anchored to DFC strands within a dispersed GC matrix [8]. Furthermore, immunofluorescence studies have revealed cell type-specific staining patterns, while FC/DFC markers typically form discrete puncta in common cell lines (e.g., MCF-7, U-251), they occasionally coalesce into larger spherical structures in other cellular contexts [9].
According to the Human Protein Atlas (https://www.proteinatlas.org/humanproteome/subcellular/nucleoli, accessed on 1 June 2025), approximately 7% (n = 1461 proteins) of the human proteome is localized to the nucleolus, including key structural components such as fibrillarin (FBL) in the DFC and nucleophosmin (NPM) in the GC [6,10]. Notably, 89% (n = 1299) of these nucleolar proteins exhibit multiple localization in other cellular compartments, particularly the cytoplasm and mitochondria. Proteins displaying similar subcellular distribution patterns frequently demonstrate functional convergence [9]. Despite significant advances in ultrastructural characterization, the precise subnucleolar distribution and functional specialization of nucleolar proteins remain incompletely elucidated. Furthermore, the nucleolus harbors transcriptionally active rRNA genes (rDNA) along with their associated transcriptional machinery, including RNA polymerase I (Pol I), protein kinases, phosphatases, and methyltransferases [7,11,12,13]. While hundreds of nucleolar proteins involved in transcriptional regulation and precursor rRNA (pre-rRNA) processing are precisely localized within the three canonical nucleolar subdomains [6], the functional consequences of their spatial organization for efficient pre-rRNA maturation remain poorly understood. A seminal study by Chen and colleagues, employing high-resolution live-cell microscopy, identified a previously unrecognized substructure, a 200-nm thick peripheral region of the DFC (PDFC) [14]. This compartment was demonstrated to be enriched with at least 12 proteins and was confirmed through three-dimensional modeling as an evolutionarily conserved substructure within each FC-DFC unit. Importantly, functional analyses established the PDFC’s essential role in rRNA maturation [14]. These findings not only provide crucial insights into the functional significance of nucleolar protein localization but also suggest the existence of additional, yet-to-be-discovered functional subdomains within the nucleolus.
3. Mechanisms of Nucleolus Formation
In addition to conventional membrane-bound organelles including secretory vesicles, the Golgi apparatus, and the endoplasmic reticulum, eukaryotic cells contain numerous membrane-less ribonucleoprotein (RNP) assemblies [15]. These comprise both nuclear compartments (e.g., nucleoli and Cajal bodies) and cytoplasmic structures (e.g., stress granules and processing bodies). Collectively referred to as biomolecular condensates [16], these membrane-delimited organelles maintain discrete boundaries while exhibiting dynamic internal organization, thereby enabling spatial segregation of specific proteins and RNAs. Notably, despite their lack of lipid membranes, these RNP condensates demonstrate functional equivalence to traditional organelles in regulating biochemical reaction efficiency through selective molecular partitioning [17]. An emerging paradigm suggests that many RNPs possess liquid-like properties and undergo formation through liquid-liquid phase separation (LLPS) [18,19,20,21]. This physicochemical process, whereby a homogeneous solution spontaneously separates into coexisting dense and dilute phases, is driven by multivalent macromolecular interactions. LLPS has been increasingly recognized as a fundamental mechanism governing the biogenesis of intracellular condensates [16,22]. Significantly, phase-separated compartments have been demonstrated to participate in multiple essential cellular processes, including higher-order chromatin organization, transcriptional regulation, selective autophagy of misfolded proteins, signal transduction complex assembly, and coordination of cytoskeletal dynamics involving both actin filaments and microtubules [16].
The nucleolus is assembled through LLPS, forming distinct liquid phases or domains characterized by differential surface tensions that prevent mixing [23]. Notably, the GC is enriched with negatively charged proteins such as NPM, which undergoes droplet formation via phase separation upon RNA binding. This liquid-like property is postulated to facilitate its functional role in ribosome biogenesis [23]. Emerging evidence demonstrates that intrinsically disordered proteins and low-complexity domain-containing proteins drive nucleolar assembly through phase transitions [16,24]. Consistent with this notion, multiple nucleolar proteins, including FBL and NPM, have extensive intrinsically disordered regions (IDRs) [6,9,23], which are enriched in charged domains essential for LLPS [6,25,26]. Of particular significance, many nucleolar proteins possess glycine-arginine-rich domains [27], wherein arginine residues alongside lysine constitute the core determinants of nucleolar localization signals [28]. These domains have been shown to promote the spontaneous formation of proteinaceous droplets both in vitro and in vivo [29,30]. Seminal work by Feric et al. demonstrated that purified nucleolar proteins (NPM, FBL, and POLR1E) undergo immiscibility-driven phase separation, recapitulating nucleolar stratification, wherein differential surface tensions sustain its multilayered architecture [15]. Domain-specific analyses further elucidated that IDRs mediate protein condensation into phase-separated droplets. Beyond proteinaceous components, rRNA [31] and long non-coding RNAs (lncRNAs) [32] have been implicated in nucleolar LLPS and structural organization. Collectively, these findings underscore the critical role of protein-protein and protein-RNA interactions in cellular LLPS, wherein molecular mixtures of proteins and RNA substrates undergo phase separation at saturation concentrations [16]. Additional regulatory factors, including pH, temperature, and post-translational modifications (PTMs), have also been reported to modulate LLPS [16,33,34]; however, their precise mechanistic contributions to nucleolar microenvironments remain poorly understood. Recent advances by Ye et al., employing fluorescence lifetime imaging microscopy with environment-sensitive fluorophores, revealed that micro-polarity gradients are essential for multilayered condensate formation [35]. Specifically, the GC layer exhibits higher micro-polarity than the DFC, with micro-polarity variations directly driving structural transitions [35]. Notably, actinomycin D (Act D) treatment was found to induce GC depolarization, concomitant with DFC hyperpolarization and subsequent nucleolar cap formation [35]. This work establishes micro-polarity as a previously unrecognized yet critical determinant of membraneless organelle organization and functionality.
4. The Functions of the Nucleolus
Despite its membrane-less architecture, the nucleolar protein network is tightly regulated during cellular growth and proliferation [12]. Functioning as the central hub for ribosome biogenesis, the nucleolus orchestrates critical processes including cell cycle progression, cellular senescence, and stress responses [36,37]. In addition to these canonical roles, it contributes to signal recognition particle assembly, telomerase regulation, and exerts tumor-suppressive or oncogenic functions [36,37]. Notably, the nucleolar proteome undergoes dynamic remodeling via continuous protein exchange, thereby adapting to fluctuating cellular demands and environmental stresses [38]. Of particular interest, numerous nucleolar functions are mediated by the selective sequestration or release of cell cycle-regulating transcription factors [37,39,40], highlighting its remarkably sophisticated regulatory capacity in the absence of membrane delineation.
4.1. Participate in the Synthesis and Assembly of Ribosomes
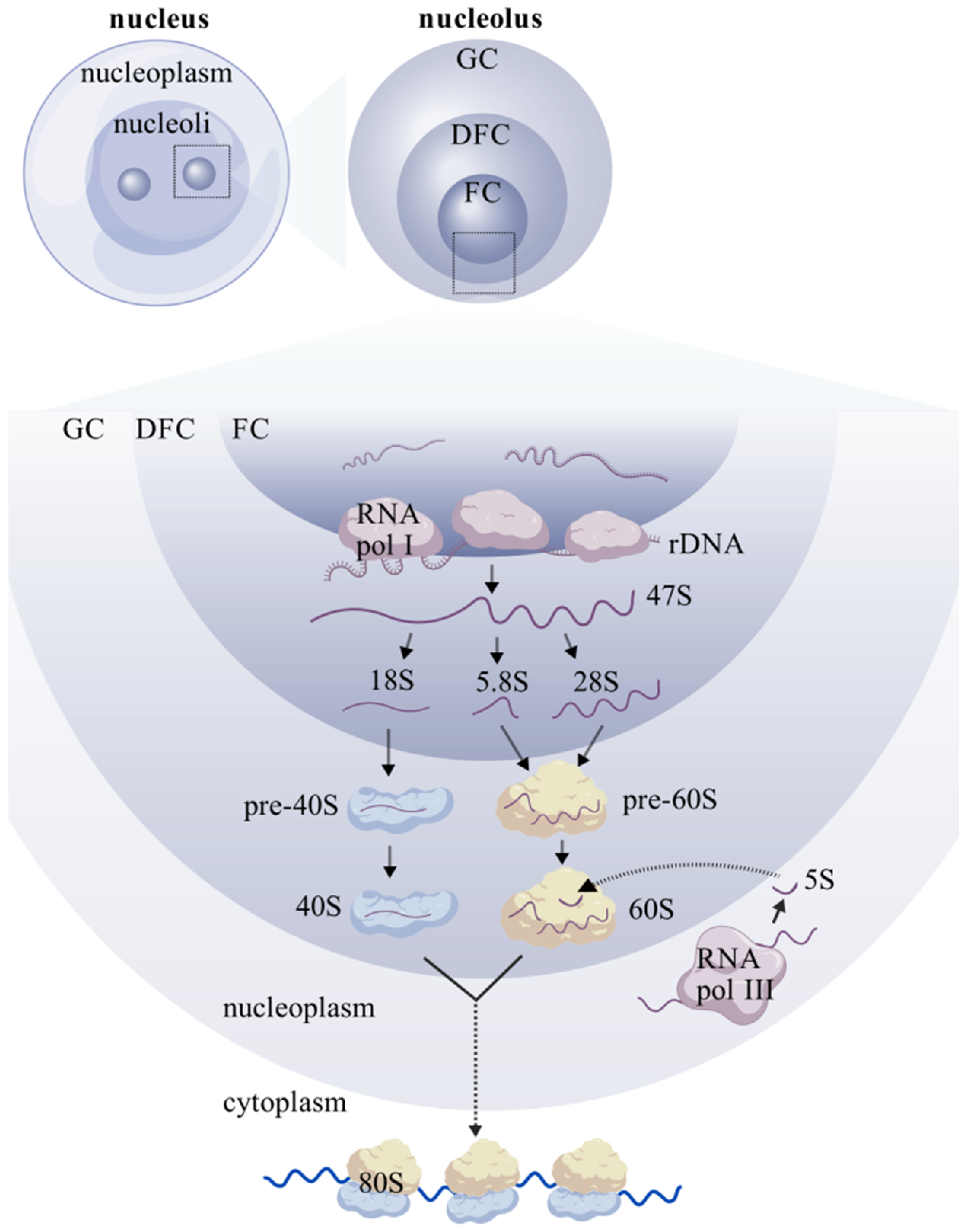
Extensive studies have established that the precise microscopic architecture of the nucleolus is intrinsically linked to its role in regulating ribosomal biogenesis. The FC serves as the primary repository for NORs, which have rDNA sequences. During active transcription, rDNA relocates from the FC core to the FC-DFC interface, where RNA Pol I catalyzes the synthesis of a 47S precursor rRNA (pre-rRNA) [7,41]. Subsequent processing occurs through a spatially organized cascade: the pre-rRNA undergoes sequential cleavage and post-transcriptional modifications within the DFC before being transported to the GC for final maturation. This process yields three critical rRNA species—5.8S, 18S, and 28S rRNA. Notably, while 5.8S and 28S rRNA (along with 5S rRNA derived by RNA pol III [41]) constitute the large (60S) ribosomal subunit, 18S rRNA exclusively forms the small (40S) subunit [5,7]. These subunits are subsequently exported to the cytoplasm, where they assemble into functional 80S ribosomes capable of mRNA binding and protein synthesis [42] (Figure 1). The dynamic nature of nucleolar proteins reflects cellular demands, with ribosomal biogenesis being particularly sensitive to cell cycle progression [11].
As ribosome function directly determines translational capacity [43,44,45,46], perturbations in ribosomal biogenesis can have profound consequences. This relationship is exemplified in ribosomopathies, where Mills and Green demonstrated that mutations in ribosomal proteins (RPs) frequently lead to functional impairments and reduced global protein synthesis [44]. Their comprehensive analysis supports two non-mutually exclusive mechanistic hypotheses. First, ribosomal dysfunction may preferentially affect mRNA-specific translational control, with certain cell types exhibiting heightened sensitivity. This vulnerability stems from differential mRNA dependence on ribosome concentration—transcripts with inefficient translation initiation are particularly susceptible to ribosomal perturbations [44]. Supporting evidence includes the observed hypersensitivity of reticulocytes and platelets to ribosomopathies, likely due to unique aspects of their ribosome recycling and rescue pathways [47]. Disruptions in these specialized mechanisms can profoundly impact ribosome homeostasis and consequently, global gene expression patterns. Alternatively, emerging evidence suggests that tissue-specific ribosome heterogeneity—whether through variable core components, differential protein composition, or distinct PTMs—may facilitate specialized translational programs [44]. This paradigm challenges the traditional view of ribosomes as uniform molecular machines, instead positing that ribosomal specialization could contribute to the tissue-specific manifestations observed in ribosomopathies.
4.2. Regulation of the Cell Cycle and Apoptosis
Beyond its canonical role in ribosomal biogenesis, the nucleolus has been implicated in the regulation of cell cycle progression and apoptotic pathways [5,48]. In eukaryotic systems, the cell cycle comprises a tightly regulated, irreversible sequence of phases (G1, S, G2, and M), each characterized by distinct biochemical and morphological events [49]. Notably, the nucleolus undergoes cyclical disassembly and reassembly, with its structural integrity and functional capacity dynamically modulated in synchrony with nuclear remodeling. Key nucleolar transitions are orchestrated at critical cell cycle checkpoints, as delineated below:
(1) S/G2 phase transition: Following the completion of DNA replication in S phase, cells enter G2, a preparatory phase preceding chromosomal segregation in mitosis [48].
(2) M phase initiation: The accumulation of cyclin B1-cyclin-dependent kinase 1 (CB1-CDK1) complexes induces hyperphosphorylation of the RNA Pol I initiation machinery, resulting in nucleolar deformation and progressive volumetric reduction concomitant with chromatin condensation [50].
(3) M phase progression: As mitosis proceeds, rRNA transcripts are dispersed, transcriptional silencing ensues, and chromatin undergoes maximal compaction, culminating in nuclear envelope breakdown and nucleolar dissolution [50].
(4) M phase termination: Upon mitotic exit, the decline in CB1-CDK1 activity facilitates the resumption of rRNA synthesis and nucleolar reformation via the fusion of prenucleolar bodies at NORs [5,51].
(5) G1 checkpoint surveillance: This stringent regulatory mechanism arrests the cell cycle upon detection of DNA lesions or aberrant spindle assembly, thereby enabling repair processes or triggering programmed cell death in metazoans [48].
Although fully functional nucleoli are reassembled during G1 phase, their structural organization and functional capacity exhibit remarkable dynamism throughout interphase. Notably, the stage-specific recruitment of various nucleolar-associated proteins suggests an active regulatory role in cell cycle progression [52]. Among the most dynamic alterations observed are PTMs, which serve as critical regulators of diverse cellular processes. Intriguingly, the nucleolus itself participates in modulating specific PTMs, including protein polymerization and phosphorylation events [53]. Reversible protein phosphorylation, in particular, represents a fundamental regulatory mechanism governing pivotal cell cycle transitions. Substantial evidence demonstrates that nucleoli contribute significantly to the phosphoregulation of cell cycle components. For instance, nucleolar-mediated phosphorylation dynamics have been shown to directly influence cyclin-dependent processes. A compelling illustration of this regulatory paradigm is observed in Saccharomyces cerevisiae, where the nucleolus-localized protein phosphatase Cdc14 executes critical control over mitotic exit through dephosphorylation of Cdh1, the activator of mitotic cyclin degradation [53]. This mechanism effectively promotes the activation of cyclin-dependent kinases, thereby facilitating proper cell cycle progression.
4.3. Participate in Nucleolar Stress Response
The precise molecular definition of nucleolar stress remains to be comprehensively elucidated within the scientific community. Originally conceptualized as perturbations disrupting ribosomal biogenesis homeostasis that subsequently activate cellular stress responses, this phenomenon has been variously designated as “ribosomal stress” or “ribotoxic stress” [54,55]. Canonical inducers encompass RNA pol I inhibitors such as Act D [56] or dysregulated expression of nucleolar proteins that compromise ribosomal function [57]. Contemporary perspectives posit nucleolar stress as comprising both morphological and functional aberrations of the nucleolus induced by diverse stressors, ultimately culminating in cellular homeostasis disruption through either p53-dependent or -independent signaling cascades. Notably, Cohen et al. employed complementary fluorescence imaging modalities to systematically quantify translocation dynamics of >1000 endogenously labeled proteins in living cells following topoisomerase I (topo I) inhibition [58]. Their seminal work demonstrated that the predominant population of drug-responsive translocating proteins were nucleolar constituents, with a pronounced reduction in topo I nucleolar intensity being observed within 2 min post-treatment [58]. These findings strongly implicate the nucleolus as both a primary sensor of transcriptional impairment and an active modulator of early stress response pathways.
4.3.1. Changes in Nucleoli under Stress Conditions
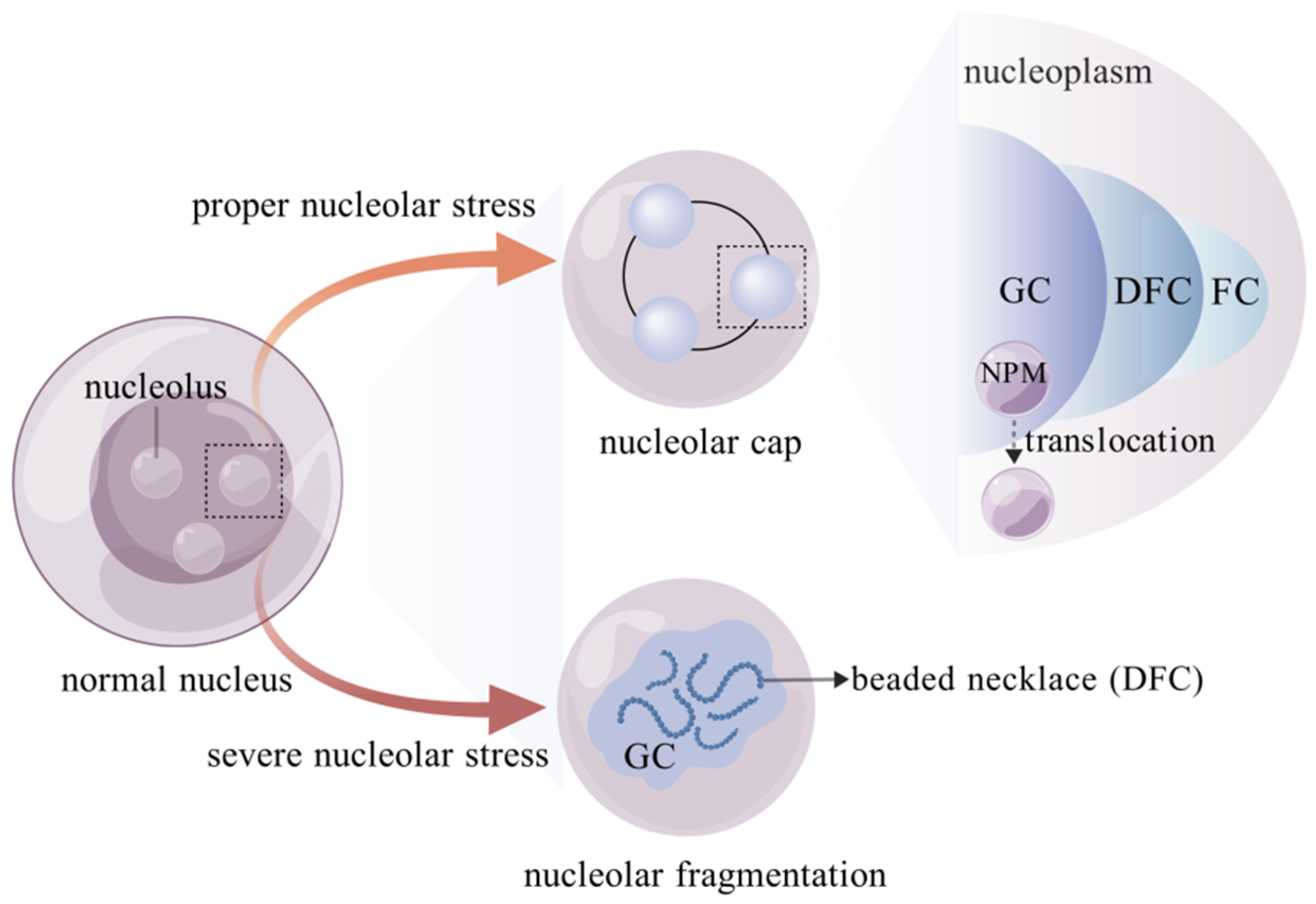
Proteomic analyses have elucidated a spectrum of dynamic nucleolar responses to diverse stress stimuli, including hypoxia, oxidative stress, transcriptional inhibition (e.g., by Act D), viral infections, and DNA damage (e.g., UV irradiation or etoposide-induced topoisomerase II inhibition), all of which perturb nucleolar microstructure and biological function [12,59,60,61,62,63]. Notably, distinct stress modalities differentially impact ribosomal subunit biogenesis and cellular growth, consistently correlating with profound alterations in nucleolar organization and proteomic composition [5]. High-resolution proteomic and electron microscopic studies demonstrate that stress-induced nucleolar segregation involves the physical separation of the FC and DFC from the GC, forming three distinct parallel architectures [6,7]. In this configuration, the FC establishes discrete caps associated with corresponding GC centrosomes, constituting the characteristic “nucleolar cap” structure [7,64] (Figure 2). Conversely, under conditions of ribosomal protein depletion that induce severe nucleolar fragmentation, DFC adopts a beaded necklace-like morphology embedded within the expanded GC regions [11] (Figure 2). It is crucial to distinguish nucleolar segregation from fragmentation events: while the former occurs following RNA Pol I inhibition [11], the latter predominantly results from RNA Pol II or protein kinase inhibition [58], manifesting as the FC disintegration and the formation of nucleolar foci alongside residual caps [65]. Furthermore, viral infections elicit specific nucleolar morphological alterations, including nucleolar hypertrophy [66,67]. These observations collectively establish that nucleolar number, architecture, and morphology serve as sensitive indicators of cellular stress response states [68,69]. For instance, heightened protein synthesis demands trigger nucleolar proliferation and volumetric expansion to augment ribosomal biogenesis capacity [70]. The nucleolar proteome exhibits remarkable dynamism, encompassing far more constituents than those strictly required for ribosome production [70]. Under specific stress conditions, prominent nucleolar proteins (particularly NPM1 [65,71]) undergo nucleolar-to-cytoplasmic translocation [10,46]. Yang et al. pioneered the demonstration of nucleolar oxidation as a universal stress response through single-cell live imaging coupled with redox biosensors [72]. Their work revealed that oxidative stress induces S-glutathionylation of NPM1 at cysteine 275, precipitating its dissociation from nucleolar nucleic acids [72]. Although RNase or DNase treatment may partially contribute to NPM1 displacement, this effect is negligible compared to oxidation-mediated regulation [72]. Nevertheless, the precise mechanisms governing nucleolar protein relocalization remain incompletely characterized and warrant further investigation.
4.3.2. Changes in LLPS during Nucleolar Stress
The nucleolus has been demonstrated to actively participate in LLPS processes, contributing to the formation of biomolecular condensates [23,73]. Notably, Frottin et al. revealed that the GC exhibits intrinsic chaperone-like activity, enabling the selective sequestration of nuclear and cytoplasmic proteins, including misfolded protein aggregates [10,23]. Under nucleolar stress conditions, misfolded proteins are recruited into the liquid-like GC matrix, where they undergo transient interactions with nucleolar proteins such as NPM. These dynamic associations significantly reduce protein mobility within the GC phase, thereby preventing irreversible protein aggregation [6]. Upon stress resolution, the dissociation of misfolded proteins from the GC compartment is precisely regulated by heat shock protein 70 chaperones, which facilitate either protein refolding or proteasomal degradation [23]. This quality control mechanism enables nucleolar proteins to regain their native functional states [23]. However, the nucleolar protein sequestration capacity is physiologically constrained. Prolonged stress exposure induces a deleterious liquid-to-solid phase transition, characterized by the formation of amyloidogenic aggregates [6,23]. These pathological transformations progress through distinct stages: initially forming granular aggregates that, if not efficiently cleared by the ubiquitin-proteasome system [10], evolve into fibrillar structures [74], as quantitatively demonstrated by fluorescence recovery after photobleaching, droplet coalescence assays, and microrheological analyses. Such aberrant phase transitions have been implicated in the pathogenesis of various neurodegenerative disorders [74,75,76,77]. Complementarily, low-dose Act D treatment provides mechanistic insights into nucleolar stress responses. While primarily inhibiting ribosomal RNA synthesis, Act D simultaneously induces structural reorganization of nucleolar compartments, culminating in the characteristic “nucleolar cap” formation through DFC-GC separation [6]. This nucleolar perturbation promotes the accumulation of stable protein aggregates in the nucleoplasm, underscoring the critical role of nucleolar LLPS properties in maintaining proteostasis [6]. The resulting aggregates exhibit pronounced cytotoxicity by sequestering functional proteins and disrupting normal cellular physiology. Similarly, in neurodegenerative pathologies such as amyotrophic lateral sclerosis and frontotemporal dementia, C9ORF72 hexanucleotide repeat expansions generate charged dipeptide repeat proteins (DPRs) with high rRNA-binding affinity [78,79]. These pathogenic DPRs competitively displace NPM from rRNA complexes, thereby destabilizing the GC architecture [78,79]. Consequently, this disruption of nucleolar LLPS dynamics impairs ribosomal biogenesis and global protein synthesis, ultimately triggering cell death pathways [78,79].
4.3.3. Signaling Pathways Involved in Nucleolar Stress
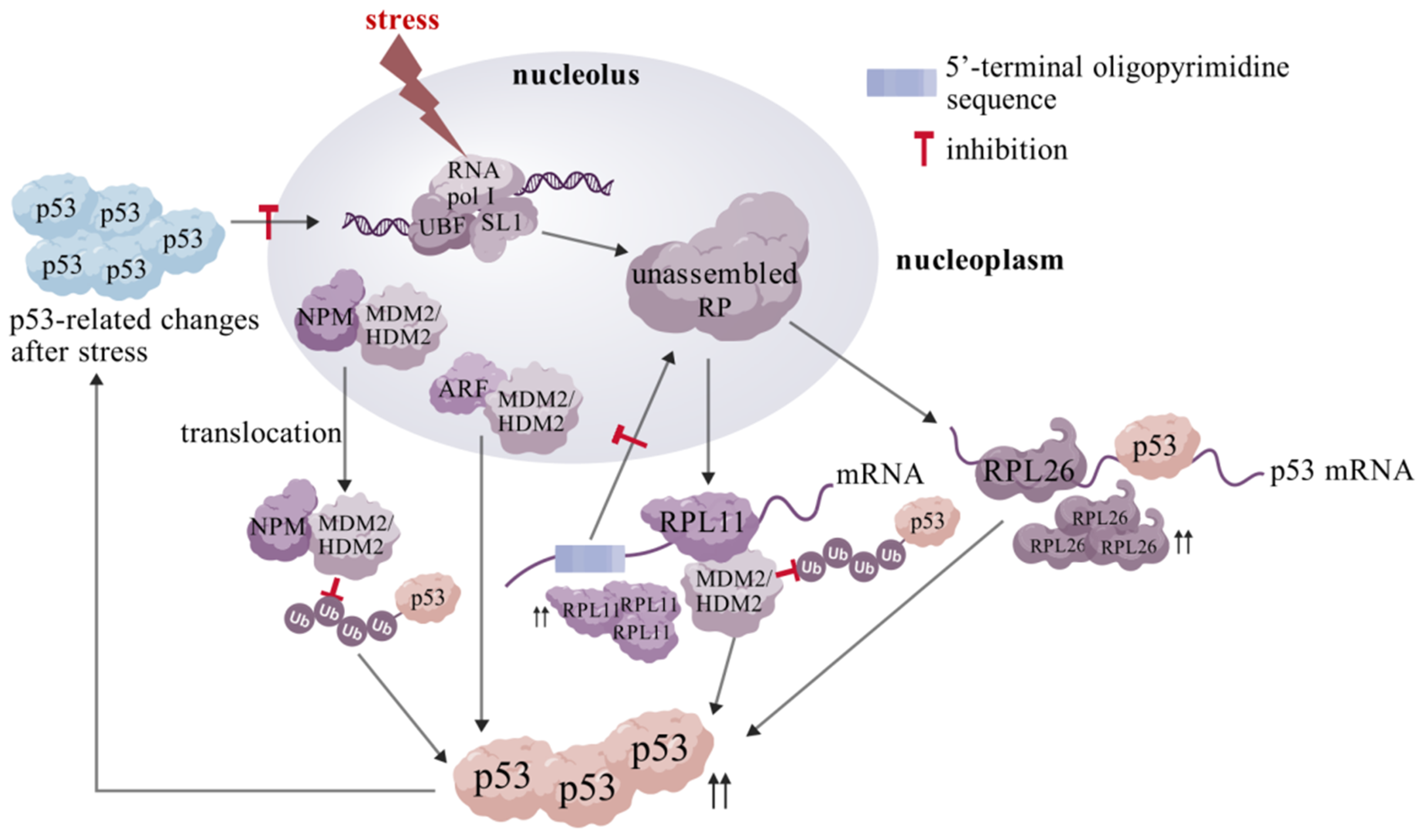
Emerging evidence demonstrates that nucleoli function as sophisticated stress sensors during cellular stress responses [65,80]. Nucleolar stress induced by aberrant rRNA transcription compromises nucleolar integrity, prompting the translocation of nucleolar proteins including NPM and nucleostemin (NS) to the cytoplasm. These relocalized proteins subsequently interact with diverse signaling molecules, modulating their stability and activity [23,81,82,83,84,85], thereby either activating or suppressing various signaling cascades [86,87]. The p53 pathway represents the most extensively characterized stress response mechanism downstream of nucleolar stress [70,88,89]. Within the nucleolar context, p53 primarily functions to upregulate RNA pol II-dependent transcription of target genes including p21, Bax, Puma, and Noxa [57]. Conversely, p53 simultaneously suppresses RNA pol I activity by disrupting the SL1-UBF interaction, consequently attenuating ribosomal subunit production [54]. Mechanistically, p53 regulation during nucleolar stress operates through three distinct paradigms [70]: (1) protein-protein interaction networks, (2) nucleolar protein translocation events, and (3) transcriptional/translational modulation (Figure 3).
During nucleolar stress, ribosomal biogenesis is impaired, leading to the accumulation and subsequent cytoplasmic release of unassembled RPs such as RPL5, RPL11, and RPL23 [42]. These RPs then bind MDM2 (murine double minute 2) or HDM2 (human homolog), the principal E3 ubiquitin ligase responsible for p53 ubiquitination and degradation [55]. This interaction inhibits MDM2/HDM2 ubiquitin ligase activity, thereby stabilizing p53 protein levels [65,90]. Notably, translational regulation contributes significantly to this stress response. The mRNAs encoding RPL11 and numerous other RPs contain 5′-terminal oligopyrimidine motifs, enabling their selective translation during global protein synthesis inhibition [91]. This translational preference may account for observed increases in nucleoplasmic RPL11 levels and consequent HDM2 inhibition [92]. Similarly, stress-induced dissociation of RPL26 from 60S ribosomal subunits elevates free RPL26 concentrations, which enhances p53 mRNA translation through direct binding to its 5′ untranslated region [93,94]. Beyond RPs, the nucleolar protein p19ARF (murine)/p14ARF (human), a highly basic, arginine-rich protein, stabilizes and interacts with the central acidic domain of MDM2/HDM2 to prevent p53 degradation [5,65,95]. Classical stress stimuli also induce NPM translocation, which exhibits dual regulatory functions: direct MDM2/HDM2 binding and inhibition [10,96], and C-terminal domain-mediated enhancement of p53 transcriptional activity [97]. Furthermore, nucleolar stress activates cell cycle checkpoints through ATM/ATR-dependent p53 phosphorylation [98,99,100], or modulates ribosomal biogenesis via the mTOR signaling pathway [101,102]. The nucleolar stress-p53 axis integrates diverse cellular processes including senescence, apoptosis, autophagy, and differentiation [103,104,105,106], underscoring its pleiotropic role in stress adaptation and cellular homeostasis.
Additionally, emerging evidence demonstrates that nucleoli and nucleolar stress participate in diverse regulatory mechanisms independent of canonical p53 signaling [42]. Notably, stress-induced translocation of nucleolar proteins to extranucleolar compartments (cytoplasm or nucleoplasm) exerts effects on key cellular regulators. These relocalized proteins have been shown to: (1) directly inhibit transcriptional regulators including MYC [107], E2F-1 [108], and HIF-1α [109,110], (2) suppress metabolic mediators such as PPAN [111,112], (3) attenuate inflammatory signaling through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway modulation [113,114,115], and (4) impair cell cycle progression via CDK4/6 inhibition [116]. Conversely, certain translocated nucleolar proteins can activate pro-apoptotic factors such as Bax, thereby triggering cell cycle arrest and/or programmed cell death [42]. This bidirectional regulatory capacity highlights the nucleolus’ role as a multifaceted stress-responsive hub that orchestrates diverse cellular outcomes through both p53-dependent and -independent mechanisms.
5. The Role of Nucleolar Stress in Cardiovascular Diseases (CVDs)
Accumulating evidence has elucidated the pivotal role of nucleolar dynamics in cellular stress responses and the pathogenesis of human diseases, with nucleolar stress being mechanistically linked to various pathological conditions including neoplastic transformation, neurodegenerative disorders, and aging processes [117,118,119]. Notably, emerging studies have demonstrated a significant association between nucleolar dysfunction and the development of cardiovascular pathologies [87,120]. Experimental investigations by Avitabile et al. revealed that α1-adrenergic receptor agonist treatment in neonatal rat cardiomyocytes induced marked nucleolar remodeling in border zone (BZ) cells of myocardial infarction (MI), characterized by nucleolar hypertrophy and morphological irregularities [80]. These pathological alterations were subsequently corroborated in human MI specimens, where concomitant cytoplasmic translocation of NS and NPM was observed [80], and at the same time, nucleolar stress exacerbates p53-mediated cell death. Furthermore, cardiotoxic chemotherapeutic agents such as Act D and doxorubicin (DOX) have been shown to induce nucleolar decondensation and mislocalization of nucleolar stress sensor proteins in cardiomyocytes [80,121,122,123], therefore leading to myocardial cell apoptosis. Clinical observations in patients with chronic ischemic cardiomyopathy and dilated cardiomyopathy (DCM) have identified significant nucleolar abnormalities, including: (1) nucleolar enlargement, (2) ultrastructural reorganization featuring reduced granular components with corresponding fibrillar component expansion, and (3) upregulated ribosomal biogenesis activity [124] (Figure 4). Importantly, sustained nucleolar hypertrophy accompanied by the increased FC, DFC, and peri-nucleolar chromatin accumulation serves as a hallmark of persistent nucleolar stress [124].
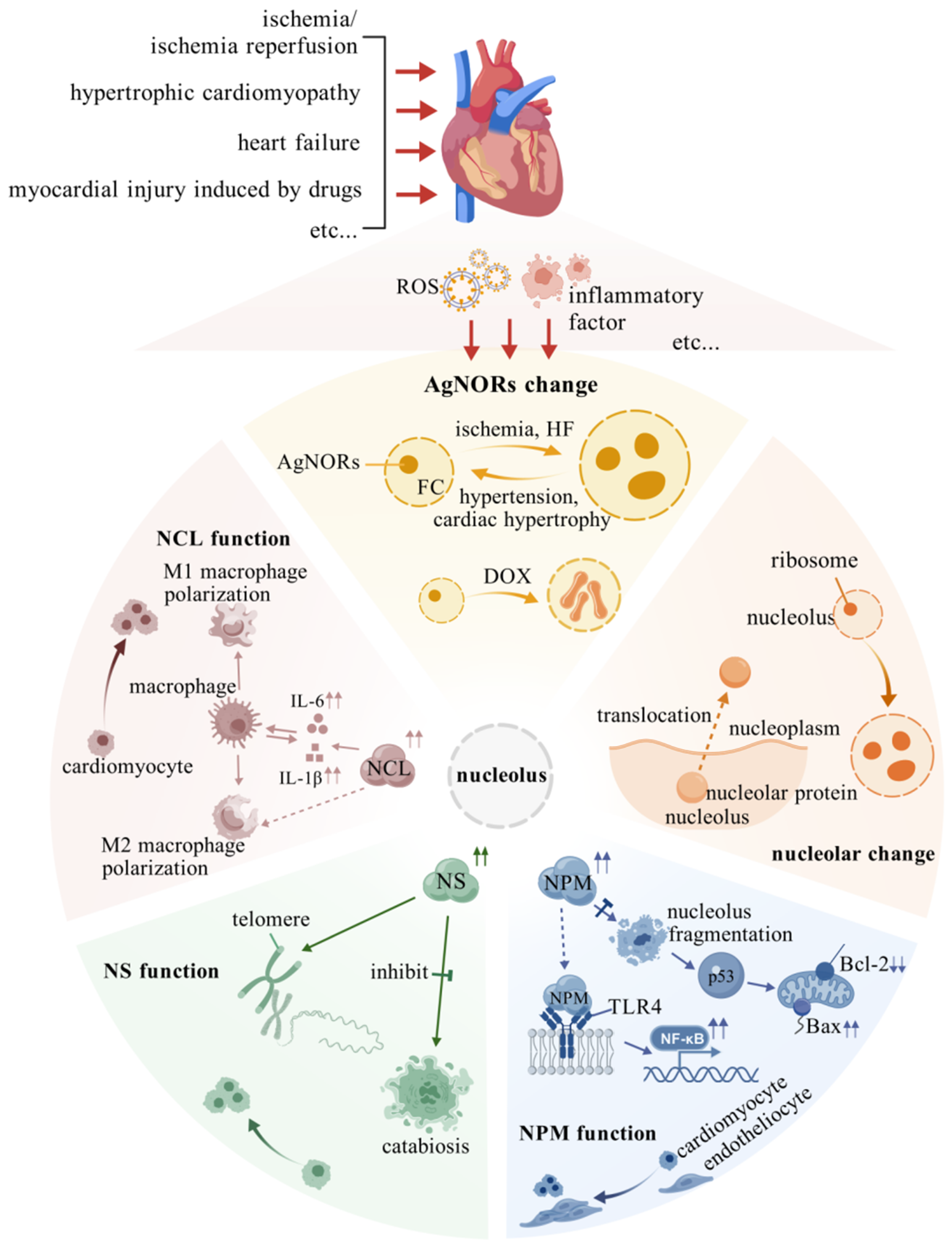
However, the Sussman research group established that MI induces rapid upregulation of nucleolar proteins, representing an early stress response event [87]. Their findings demonstrate that nucleolar stress initiates both the transcriptional activation and subcellular redistribution of cardioprotective nucleolar proteins. Among these, NS, NPM and nucleolin (NCL) have been most extensively characterized [87]. These proteins exhibit dual functionality in cardiomyocytes through both mediating cell survival pathways and concurrently activating inflammatory cascades along with facilitating cardiac tissue repair [120,125,126]. Accumulating clinical and experimental evidence positions the nucleolus as a critical regulator in cardiovascular pathogenesis, where nucleolar protein expression dynamics substantially influence cardiomyocyte fate determination [70]. Notably, as primary cellular stress sensors, nucleoli respond to injury through both structural reorganization and activation of downstream signaling networks [70,127]. In acute MI adult models, Siddiqi and colleagues made several key observations that relatively low levels of NS expression in the myocardial nucleus mainly located in the ischemic zone and BZ significantly increase due to MI [128]. Temporal analysis by western blot revealed: (1) detectable NS elevation by 24 h post-MI, (2) significant protein accumulation by 48 h, (3) peak expression at 72 h and (4) subsequent decline to baseline within 7 days [128]. Western blot quantification confirmed this biphasic expression pattern, with maximal NS levels corresponding to the critical window for myocardial repair processes [128]. However, distinct nucleolar proteins exhibit differential temporal expression patterns in response to MI-induced stress. For instance, while NCL shows an immediate reduction within 24 h post-MI [81], its expression becomes significantly elevated during the 7–28 days recovery period [129]. Current investigations have established NPM as a critical nucleolar phosphoprotein [130,131] that orchestrates cellular survival and proliferation pathways. Mechanistically, NPM is indispensable for maintaining genomic integrity through its roles in DNA repair and chromosomal stability [130,132]. NPM deficiency triggers a cascade of detrimental effects including: (1) nucleolar fragmentation, (2) inhibition of precursor RNA synthesis, and (3) induction of cardiomyocyte apoptosis [80]. Furthermore, NPM has been demonstrated to modulate NF-κB activity during endothelial cell senescence [133] (Figure 4). Intriguingly, in human cardiac mesenchymal progenitor cells, NPM undergoes autophagy-dependent secretion into the extracellular compartment [134], where it functions as an endogenous ligand for Toll-like receptor 4 (TLR4). This interaction initiates TLR4/NF-κB-mediated inflammatory signaling, potentially facilitating cardiac tissue repair processes [135] (Figure 4). Csiszar et al. have established a molecular link between NPM and atherogenesis, demonstrating that elevated NPM mRNA expression in aged rat carotid arteries and cardiac tissue correlates with enhanced NF-κB activation [133]. These findings posit NPM as a key regulator of oxidative stress responses and pro-inflammatory cascades implicated in cardiovascular aging. Complementary in vitro studies utilizing oxidized low-density lipoprotein (oxLDL)-treated human vascular endothelial cells reveal that NPM dephosphorylation is associated with both diminished proliferative capacity and exacerbated cellular dysfunction [136]. Conversely, Jiang et al. have demonstrated that NCL overexpression confers cardioprotection against hypoxia- and H2O2-induced cardiomyocyte apoptosis [81]. This protective role is further substantiated by observations in transgenic murine models, where cardiac-specific NCL overexpression attenuates ischemia-reperfusion injury, as evidenced by reduced cellular necrosis and diminished infarct size [81]. These collective findings suggest that NCL facilitates cardiomyocyte recovery following stress exposure. NCL exhibits dual cytoprotective function, such as mitigating oxidative stress-induced cell death when overexpressed while exacerbating cellular damage when depleted. Furthermore, NCL plays a vital role in modulating cytokine production, particularly interleukin (IL)-6 and IL-1β, which are critical mediators of inflammatory responses and cardiac repair processes [82,137]. Mechanistically, NCL has been shown to be indispensable for M2 macrophage polarization—a crucial immunological mechanism for myocardial tissue regeneration—as NCL depletion significantly impairs this reparative polarization [129] (Figure 4).
Nucleolar enlargement serves as a morphological hallmark of augmented protein biosynthesis, representing one of the earliest cellular adaptations to chronic stress conditions, including cardiac hypertrophy [56,138]. Siddiqi et al. systematically investigated this phenomenon in a pressure overload-induced murine model of myocardial hypertrophy, where NS expression was found to be significantly upregulated [128]. Subsequent in vitro experiments demonstrated that NS overexpression in cultured cardiac stem cells (CSCs) was functionally associated with maintenance of CSCs proliferative capacity as well as preservation of telomeric integrity [128] (Figure 4). These findings collectively suggest that NS serves as a critical regulator of cardiac proliferation and survival signaling pathways, making it as a promising molecular target for myocardial regeneration and anti-aging therapeutic strategies [80]. Concurrently, during the pathological progression from myocardial hypertrophy to heart failure (HF) in murine models, NCL has been observed to exhibit enhanced chromatin-binding affinity [139]. This increased genomic association facilitates transcriptional reprogramming events that ultimately upregulate NCL expression, thereby contributing to myocardial remodeling processes [139].
To systematically characterize nucleolar stress markers in cardiomyocytes under pathological conditions, accumulating evidence indicates that diminished nucleolar staining intensity may serve as a sensitive indicator of cellular stress and is strongly correlated with impaired cardiac function [140,141]. This phenomenon was initially documented in a clinical study demonstrating significant reduction of silver-stained nucleolar organizer regions (AgNORs) in cardiomyocytes from patients with severe ischemic heart disease complicated by HF [142]. Mechanistically, AgNORs predominantly localize to the FC layer, where specific nucleolar proteins exhibit affinity for silver nitrate staining [143]. The observed AgNORs reduction in ischemic and failing hearts is attributed to suppressed metabolic activity and attenuated rRNA biosynthesis [7,123,144]. AgNORs dynamics exhibit temporal regulation during ischemic events, for instance, rapid diminution during transient cardiac arrest while prompt restoration upon reperfusion [142,144]. Conversely, in hypertensive cardiomyopathy, NORs activity demonstrate positive correlations with key hypertrophic parameters including myocardial mass, left ventricular wall thickness, and maximal diastolic pressure [145], suggesting that nucleolar activity is augmented during compensatory cardiac hypertrophy [138]. Furthermore, DOX-induced cardiotoxicity elicits distinct AgNORs alterations characterized by both morphological enlargement and rod-like structural transformations [87]. Collectively, these findings establish NORs as highly sensitive and dynamic biomarker of cardiac pathophysiology (Figure 4).
Current evidence demonstrates that cardiomyocytes activate nucleolar stress in response to myocardial injury, unveiling its intricate pathophysiological mechanisms characterized by biphasic regulatory properties. The dichotomous outcomes of nucleolar stress are governed by critical determinants, including stress intensity, temporal dynamics, cell type and downstream signaling cascades. Under conditions of sustained or severe stress, nucleolar dysfunction exacerbates p53-dependent apoptotic signaling and enhances the release of pro-inflammatory cytokines, including TNF-α and IL-6, thereby amplifying local inflammatory cascades and compromising cardiomyocyte viability and function. Conversely, moderate or transient nucleolar stress elicits adaptive autophagy, a cytoprotective mechanism that facilitates cellular self-preservation. In this context, nucleolar stress-induced autophagy promotes the clearance of damaged organelles and misfolded protein aggregates, thereby preserving intracellular homeostasis and attenuating inflammatory mediator production through suppression of p53-mediated apoptosis [114,146] and inhibition of NF-κB signaling [133,135]. These mechanisms collectively support myocardial repair and regeneration. The temporal dynamics of nucleolar stress dictate its divergent effects: during the early phase, nucleolar stress induces transient cell cycle arrest, allowing for cellular repair and damage mitigation. However, prolonged stress ultimately overwhelms DNA damage repair mechanisms, driving cells toward apoptotic or necrotic death. Furthermore, nucleolar stress engages multiple regulatory pathways, including modulation of mRNA processing and ribosomal biogenesis, which can profoundly influence cellular metabolic states and shift the balance between survival and death. Given its pivotal role in determining cell fate, a comprehensive understanding of nucleolar stress mechanisms is essential for developing novel therapeutic interventions aimed at ameliorating myocardial injury.
6. The Role of LLPS in CVDs
Numerous studies have established that LLPS participates in diverse cellular processes, including adaptive and innate immune signaling, stress granule formation, heterochromatin organization, and transcriptional regulation [22,147]. Although there is currently no direct evidence indicating the presence of phase separation in CVDs, these LLPS-mediated mechanisms play key roles in the pathogenesis of various disorders, particularly cancer, neurodegenerative diseases, and inflammatory conditions [22,147]. Notably, emerging research indicates that LLPS dysregulation manifested through aberrant protein, nucleic acid and other biological activities, significantly contributes to cardiovascular pathophysiology [25].
For instance, Horii et al. identified vestigial-like family member 3 (VGLL3) as a mechanosensitive protein specifically expressed in cardiac fibroblasts [148]. Their work demonstrated that VGLL3 undergoes LLPS through its glutamic acid-rich IDRs, thereby promoting cardiac fibrosis [148]. Complementarily, Xie and his colleagues revealed that arachidonate 5-lipoxygenase facilitates the nuclear translocation of RUNX family transcription factor 2 (Runx2) in an enzyme activity-independent manner [149]. This interaction enhances Runx2 LLPS within the nucleus, subsequently activating EGFR and MAPK signaling pathways in cardiomyocytes. Importantly, pharmacological inhibition of Runx2 phase separation was shown to attenuate pathological cardiac remodeling in heart failure models [149]. Furthermore, Jiang et al. found hematopoietic progenitor kinase 1-interacting protein of 55 kDa (HIP-55) as a potent phase-separating protein whose activity is regulated by AKT-mediated phosphorylation at residues S269 and T291 [150]. Under conditions of chronic sympathetic overactivation, reduced phosphorylation at these sites disrupts HIP-55 phase separation capacity, leading to the formation of insoluble aggregates and consequent loss of cardioprotective function in HF [150]. The dynamic phase separation behavior of HIP-55 has been demonstrated to critically regulate β-adrenergic receptor signaling by attenuating excessive activation of the P38/MAPK pathway [150]. Complementarily, Schneider et al. noted that aberrant phase separation of RBM20 mutants leads to pathological accumulation of RNP particles within the sarcoplasm [151]. These mislocalized RNP complexes, containing mutant RBM20, preferentially accumulate at Z-disc regions of the cytoskeletal network, where they impair microtubule-mediated transport of genetic material [151]. Moreover, dysregulated RNP particles sequester actin α1 at phase boundaries, disrupting physiological actin polymerization dynamics and thereby contributing to the pathogenesis of DCM [151]. Additionally, messenger RNPs (mRNPs) are transported along microtubules to sites of myofibril synthesis in cardiomyocytes, where they not only mediate cardiac hypertrophy but may also participate in intercalated disc formation during this process [152]. In the context of atherosclerosis, LLPS has been implicated in both autophagy impairment and pro-inflammatory responses induced by oxLDL. Li et al. provided direct evidence that macrophage LLPS participates in atherogenesis, highlighting its role in vascular pathology [153].
Notably, while protein-RNA interactions represent a major driver of LLPS, the precise mechanisms by which LLPS governs nucleolar assembly in CVDs remain poorly understood and warrant further exploration. We propose that under CVDs conditions, IDRs within nucleolar proteins undergo a disorder-to-order transition, accompanied by mutations in key charged residues of IDRs. The sequence composition, length, and post-translational modifications of these IDRs critically influence the biophysical properties and functional outcomes of LLPS-driven condensates, ultimately disrupting nucleolar architecture and impairing the proper translation and folding of nucleolar-associated proteins. Furthermore, specific rRNAs, mRNAs, and lncRNAs may actively modulate nucleolar phase separation, thereby contributing to aberrant nucleolar organization in CVDs.
7. Prospects
With advancing exploration of cellular microstructure and function, the nucleolus has emerged as a critical regulator in cardiovascular pathophysiology, unveiling novel avenues for both fundamental research and clinical translation. From a basic research perspective, it is imperative to decipher the precise regulatory mechanisms governing nucleolar signaling pathways under stress conditions, elucidate how the nucleolus maintains or remodels its structural integrity through processes such as LLPS during pathological states, and systematically characterize the molecular cascades through which nucleolar dysfunction precipitates cardiomyocyte pathology through leveraging multi-omics approaches encompassing transcriptomics, proteomics, post-translational modification profiling, and metabolomics. Notably, from an epigenetic standpoint, nucleolar-associated molecules hold substantial promise as clinically actionable biomarkers, potentially enabling early disease detection with high sensitivity and specificity. Such advances would facilitate precise screening and risk stratification for diverse cardiovascular disorders.
While clinical applications of nucleolar biomarkers in CVDs remain unexplored, emerging evidence suggests their considerable diagnostic and therapeutic potential. We propose several promising nucleolar-related biomarkers for CVDs management. Primarily, dysregulated expression of nucleolar proteins observed across various CVDs could serve as direct histological markers in tissue biopsies. Additionally, given that nucleolar stress impairs ribosome biogenesis, quantification of pre-rRNA or circulating free RNPs complexes may provide sensitive indicators of cardiomyocyte stress. Furthermore, alterations in microRNAs and lncRNAs that regulate nucleolar function may serve as indirect yet accessible signatures of nucleolar dysfunction. However, significant challenges remain: the ubiquitous role of nucleolar components in fundamental cellular processes necessitates development of cardiovascular-specific detection methods, while standardization and reproducibility of nucleolar biomarker assays require substantial optimization before clinical translation.
Current research on nucleolar-targeted therapeutics remains predominantly focused on oncology, virology, and neurodegenerative disorders, with comparatively limited investigation in CVDs. Nevertheless, certain pharmacological agents may indirectly influence cardiovascular pathophysiology through modulation of nucleolar stress responses, ribosome biogenesis, nucleolar protein dynamics, or antibodies targeting critical signaling molecules. Given the distinct mechanisms underlying nucleolar pathology across disease states, we postulate that CVDs-specific nucleolar interventions would likely prioritize direct activation of ribosome biogenesis pathways, enhancement of rRNA transcription or processing efficiency, and optimization of translational efficiency to maximize nucleolar functional capacity. Spatiotemporal analyses have demonstrated that pretreatment with the antioxidant N-acetylcysteine effectively prevents stress-induced NPM1 nucleocytoplasmic translocation [65]. Given that nuclear oxidative stress represents a conserved response to diverse cellular insults, we postulate that pharmacological antioxidants may similarly preserve nucleolar architectural integrity. By maintaining proper nucleolar compartmentalization, such interventions could prevent aberrant interactions between nucleolar proteins and extranucleolar components, potentially mitigating adverse cardiovascular outcomes. This stabilization of nucleolar spatial organization may represent a novel therapeutic strategy for cardiovascular protection. Furthermore, considering the emerging role of LLPS in nucleolar organization, two potential therapeutic avenues we may assume. First, for pathological LLPS exacerbating disease progression, development of short peptide inhibitors that competitively bind to phase separation domains or interact with key LLPS-associated proteins could attenuate aberrant protein aggregation. Second, for physiologically relevant phase separation, pharmacological modulation of droplet microenvironment or stabilization of key proteins may restore homeostatic LLPS dynamics.
Above all, the development of nucleolus-targeted pharmacological agents represents a transformative frontier. By precisely modulating key nucleolar effectors implicated in cardiovascular pathogenesis, these interventions could correct aberrant nucleolar activation and function, ameliorate cardiomyocyte pathophysiology, attenuate or reverse disease progression and minimize adverse effects associated with conventional therapies. This targeted strategy would not only enhance therapeutic efficacy and safety but also improve long-term patient outcomes, ultimately propelling cardiovascular medicine toward precision and personalized care paradigms.
Author Contributions
R.X.: conceptualization, investigation, visualization and writing; X.J.: investigation, writing-reviewing and funding acquisition; Y.Z.: resources, reviewing; X.Y.: supervision; Y.X.: conceptualization, supervision, writing-reviewing and editing, project administration. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China, grant number 82400304. The APC was funded by National Natural Science Foundation of China, grant number 82400304.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Use of AI and AI-Assisted Technologies
No AI tools were utilized for this paper.
References
- 1.
Valentin, G. Repertorium für Anatomie und Physiologie; Verlag Veit & Comp.: Berlin, Germany, 1836; Volume 1, 293p.
- 2.
Heitz, E. Nukleolar und chromosomen in der gattung. Vicia Planta 1931, 15, 495–505.
- 3.
McClintock, B. The relationship of a particular chromosomal element to the development of the nucleoli in Zea mays. Z. Zellforsch. Mikrosk. Anat. 1934, 21, 294–398.
- 4.
Cerqueira, A.V.; Lemos, B. Ribosomal DNA and the Nucleolus as Keystones of Nuclear Architecture, Organization, and Function. Trends Genet. 2019, 35, 710–723.
- 5.
Boisvert, F.M.; van Koningsbruggen, S.; Navascués, J.; et al. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585.
- 6.
Lafontaine, D.L.J.; Riback, J.A.; Bascetin, R.; et al. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182.
- 7.
Hernandez-Verdun, D.; Roussel, P.; Thiry, M.; et al. The nucleolus: Structure/function relationship in RNA metabolism. Wiley Interdiscip. Rev. RNA 2010, 1, 415–431.
- 8.
Scheer, U.; Hock, R. Structure and function of the nucleolus. Curr. Opin. Cell Biol. 1999, 11, 385–390.
- 9.
Stenström, L.; Mahdessian, D.; Gnann, C.; et al. Mapping the nucleolar proteome reveals a spatiotemporal organization related to intrinsic protein disorder. Mol. Syst. Biol. 2020, 16, e9469.
- 10.
Iarovaia, O.V.; Minina, E.P.; Sheval, E.V.; et al. Nucleolus: A Central Hub for Nuclear Functions. Trends Cell Biol. 2019, 29, 647–659.
- 11.
Corman, A.; Sirozh, O.; Lafarga, V.; et al. Targeting the nucleolus as a therapeutic strategy in human disease. Trends Biochem. Sci. 2023, 48, 274–287.
- 12.
Andersen, J.S.; Lam, Y.W.; Leung, A.K.; et al. Nucleolar proteome dynamics. Nature 2005, 433, 77–83.
- 13.
Tuteja, R.; Tuteja, N. Nucleolin: A multifunctional major nucleolar phosphoprotein. Crit. Rev. Biochem. Mol. Biol. 1998, 33, 407–436.
- 14.
Shan, L.; Xu, G.; Yao, R.; et al. Nucleolar URB1 ensures 3′ ETS rRNA removal to prevent exosome surveillance. Nature 2023, 615, 526–534.
- 15.
Feric, M.; Vaidya, N.; Harmon, T.S.; et al. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697.
- 16.
Zhang, H.; Ji, X.; Li, P.; et al. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985.
- 17.
Balagopal, V.; Parker, R. Polysomes, P bodies and stress granules: States and fates of eukaryotic mRNAs. Curr. Opin. Cell Biol. 2009, 21, 403–408.
- 18.
Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732.
- 19.
Wilson, E.B. The structure of protoplasm. Science 1899, 10, 33–45.
- 20.
Li, P.; Banjade, S.; Cheng, H.C.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340.
- 21.
Kato, M.; Han, T.W.; Xie, S.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767.
- 22.
Wang, B.; Zhang, L.; Dai, T.; et al. Liquid-liquid phase separation in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 290.
- 23.
Frottin, F.; Schueder, F.; Tiwary, S.; et al. The nucleolus functions as a phase-separated protein quality control compartment. Science. 2019, 365, 342–347.
- 24.
Berry, J.; Weber, S.C.; Vaidya, N.; et al. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E5237–E5245.
- 25.
Mo, Y.; Feng, Y.; Huang, W.; et al. Liquid-Liquid Phase Separation in Cardiovascular Diseases. Cells. 2022, 11, 3040.
- 26.
Cai Z, Mei S, Zhou L; et al. Liquid-Liquid Phase Separation Sheds New Light upon Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 15418.
- 27.
Girard, J.P.; Lehtonen, H.; Caizergues-Ferrer, M.; et al. GAR1 is an essential small nucleolar RNP protein required for pre-rRNA processing in yeast. EMBO J. 1992, 11, 673–682.
- 28.
Scott, M.S.; Boisvert, F.M.; McDowall, M.D.; et al. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010, 38, 7388–7399.
- 29.
Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194.
- 30.
Nott, T.J.; Petsalaki, E.; Farber, P.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell. 2015, 57, 936–947.
- 31.
Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324.e28.
- 32.
Wu, M.; Xu, G.; Han, C.; et al. lncRNA SLERT controls phase separation of FC/DFCs to facilitate Pol I transcription. Science 2021, 373, 547–555.
- 33.
van der Lee, R.; Buljan, M.; Lang, B.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631.
- 34.
Choi, J.M.; Holehouse, A.S.; Pappu, R.V. Physical principles underlying the complex biology of intracellular phase transitions. Annu. Rev. Biophys. 2020, 49, 107–133.
- 35.
Ye, S.; Latham, A.P.; Tang, Y.; et al. Micropolarity governs the structural organization of biomolecular condensates. Nat. Chem. Biol. 2024, 20, 443–451.
- 36.
Olson, M.O.; Hingorani, K.; Szebeni, A. Conventional and nonconventional roles of the nucleolus. Int. Rev. Cytol. 2002, 219, 199–266.
- 37.
Pederson, T.; Tsai, R.Y. In search of nonribosomal nucleolar protein function and regulation. J. Cell Biol. 2009, 184, 771–776.
- 38.
Zhao, S.; Huang, D.; Peng, J. Nucleolus-localized Def-CAPN3 protein degradation pathway and its role in cell cycle control and ribosome biogenesis. J. Genet. Genom. 2021, 48, 955–960.
- 39.
Carmo-Fonseca, M. The contribution of nuclear compartmentalization to gene regulation. Cell 2002, 108, 513–521.
- 40.
Zimber, A.; Nguyen, Q.D.; Gespach, C. Nuclear bodies and compartments: Functional roles and cellular signalling in health and disease. Cell Signal. 2004, 16, 1085–1104.
- 41.
Orsolic, I.; Jurada, D.; Pullen, N.; et al. The relationship between the nucleolus and cancer: Current evidence and emerging paradigms. Semin. Cancer Biol. 2016, 37–38, 36–50.
- 42.
Maehama, T.; Nishio, M.; Otani, J.; et al. Nucleolar stress: Molecular mechanisms and related human diseases. Cancer Sci. 2023, 114, 2078–2086.
- 43.
Antony, C.; George, S.S.; Blum, J.; et al. Control of ribosomal RNA synthesis by hematopoietic transcription factors. Mol. Cell 2022, 82, 3826–3839.e9.
- 44.
Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358, eaan2755.
- 45.
Jiao, L.; Liu, Y.; Yu, X.Y.; et al. Ribosome biogenesis in disease: New players and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 15.
- 46.
Chen, D.; Huang, S. Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J. Cell Biol. 2001, 153, 169–176.
- 47.
Mills, E.W.; Wangen, J.; Green, R.; et al. Dynamic regulation of a ribosome rescue pathway in erythroid cells and platelets. Cell Rep. 2016, 17, 1–10.
- 48.
Visintin, R.; Amon, A. The nucleolus: The magician’s hat for cell cycle tricks. Curr. Opin. Cell Biol. 2000, 12, 372–377.
- 49.
Dalton, S. Linking the Cell Cycle to Cell Fate Decisions. Trends Cell Biol. 2015, 25, 592–600.
- 50.
Heix, J.; Vente, A.; Voit, R.; et al. Mitotic silencing of human rRNA synthesis: Inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO J. 1998, 17, 7373–7381.
- 51.
Shaw, P.J.; Jordan, E.G. The nucleolus. Annu. Rev. Cell Dev. Biol. 1995, 11, 93–121.
- 52.
Kroetz, M.B. SUMO: A ubiquitin-like protein modifier. Yale J. Biol. Med. 2005, 78, 197–201.
- 53.
Visintin, R.; Craig, K.; Hwang, E.S.; et al. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell 1998, 2, 709–718.
- 54.
Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000, 20, 5930–5938.
- 55.
Prives, C. Signaling to p53: Breaking the MDM2-p53 circuit. Cell 1998, 95, 5–8.
- 56.
Neuburger, M.; Herget, G.W.; Plaumann, L.; et al. Change in size, number and morphology of the nucleoli in human hearts as a result of hyperfunction. Pathol.-Res. Pract. 1998, 194, 385–389.
- 57.
Lee, J.T.; Gu, W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010, 17, 86–92.
- 58.
Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; et al. Dynamic proteomics of individual cancer cells in response to a drug. Science 2008, 322, 1511–1516.
- 59.
Al-Baker, E.A.; Oshin, M.; Hutchison, C.J.; et al. Analysis of UV-induced damage and repair in young and senescent human dermal fibro-blasts using the comet assay. Mech. Ageing Dev. 2005, 126, 664–672.
- 60.
Boisvert, F.M.; Lamond, A.I. P53-dependent subcellular proteome localization following DNA damage. Proteomics 2010, 10, 4087–4097.
- 61.
Boisvert, F.M.; Lam, Y.W.; Lamont, D.; et al. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell Proteom. 2010, 9, 457–470.
- 62.
Emmott, E.; Rodgers, M.A.; Macdonald, A.; et al. Quantitative proteomics using stable isotope labeling with amino acids in cell culture (SILAC) reveals changes in the cytoplasmic, nuclear and nucleolar proteomes in Vero cells infected with the coronavirus infectious bronchitis virus. Mol. Cell Proteom. 2010, 9, 1920–1936.
- 63.
Lam, Y.W.; Evans, V.C.; Heesom, K.J.; et al. Proteomics analysis of the nucleolus in adenovirus- infected cells. Mol. Cell Proteom. 2010, 9, 117–130.
- 64.
Shav-Tal, Y.; Blechman, J.; Darzacq, X.; et al. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell 2005, 16, 2395–2413.
- 65.
Yang, K.; Yang, J.; Yi, J. Nucleolar Stress: Hallmarks, sensing mechanism and diseases. Cell Stress 2018, 2, 125–140.
- 66.
Greco, A. Involvement of the nucleolus in replication of human viruses. Rev. Med. Virol. 2009, 19, 201–214.
- 67.
Hiscox, J.A. RNA viruses: Hijacking the dynamic nucleolus. Nat. Rev. Microbiol. 2007, 5, 119–127.
- 68.
Derenzini, M.; Trerè, D.; Pession, A.; et al. Nucleolar size indicates the rapidity of cell proliferation in cancer tissues. J. Pathol. 2000, 191, 181–186.
- 69.
Frank, D.J.; Roth, M.B. ncl-1 is required for the regulation of cell size and ribosomal RNA synthesis in Caenorhabditis elegans. J. Cell Biol. 1998, 140, 1321–1329.
- 70.
Boulon, S.; Westman, B.J.; Hutten, S.; et al. The nucleolus under stress. Mol. Cell 2010, 40, 216–227.
- 71.
Weeks, S.E.; Metge, B.J.; Samant, R.S. The nucleolus: A central response hub for the stressors that drive cancer progression. Cell. Mol. Life Sci. 2019, 76, 4511–4524.
- 72.
Yang, K.; Wang, M.; Zhao, Y. A redox mechanism underlying nucleolar stress sensing by nucleophosmin. Nat. Commun. 2016, 7, 13599.
- 73.
Jin, X.; Tanaka, H.; Jin, M.; et al. PQBP5/NOL10 maintains and anchors the nucleolus under physiological and osmotic stress conditions. Nat. Commun. 2023, 14, 9.
- 74.
Molliex, A.; Temirov, J.; Lee, J.; et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133.
- 75.
Patel, A.; Lee, H.O.; Jawerth, L.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077.
- 76.
Zhang, H.; Elbaum-Garfinkle, S.; Langdon, E.M.; et al. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell 2015, 60, 220–230.
- 77.
Murakami, T.; Qamar, S.; Lin, J.Q.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690.
- 78.
White, M.R.; Mitrea, D.M.; Zhang, P.; et al. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728.e6.
- 79.
Lee, K.H.; Zhang, P.; Joo Kim, H.J.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17.
- 80.
Avitabile, D.; Bailey, B.; Cottage, C.T.; et al. Nucleolar stress is an early response to myocardial damage involving nucleolar proteins nucleostemin and nucleophosmin. Proc. Natl. Acad. Sci. USA 2011, 108, 6145–6150.
- 81.
Jiang, B.; Zhang, B.; Liang, P.; et al. Nucleolin protects the heart from ischaemia-reperfusion injury by up-regulating heat shock protein 32. Cardiovasc. Res. 2013, 99, 92–101.
- 82.
Mariero, L.H.; Torp, M.-K.; Heiestad, C.M.; et al. Inhibiting nucleolin reduces inflammation induced by mitochondrial DNA in cardiomyocytes exposed to hypoxia and reoxygenation. Br. J. Pharmacol. 2019, 176, 4360–4372.
- 83.
Azkanaz, M.; López, A.R.; de Boer, B.; et al. Protein quality control in the nucleolus safeguards recovery of epigenetic regulators after heat shock. Elife 2019, 8, e45205.
- 84.
Liu, Y.; Wang, Y.; Yang, L.; et al. The nucleolus functions as the compartment for histone H2B protein degradation. IScience. 2021, 24, 102256.
- 85.
Hu, B.; Hua, L.; Ni, W.; et al. Nucleostemin/GNL3 promotes nucleolar polyubiquitylation of p27(kip1) to drive hepatocellular carcinoma progression. Cancer Lett. 2017, 388, 220–229.
- 86.
Sengar, A.S.; Kumar, M.; Rai, C.; et al. RGS6 drives cardiomyocyte death following nucleolar stress by suppressing Nucleolin/miRNA-21. J. Transl. Med. 2024, 22, 204.
- 87.
Hariharan, N.; Sussman, M.A. Stressing on the nucleolus in cardiovascular disease. Biochim. Biophys. Acta. 2014, 1842, 798–801.
- 88.
Donati, G.; Peddigari, S.; Mercer, C.A.; et al. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013, 4, 87–98.
- 89.
Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077.
- 90.
Núñez Villacís, L.; Wong, M.S.; Ferguson, L.L.; et al. New roles for the nucleolus in health and disease. Bioessays. 2018, 40, e1700233.
- 91.
Caldarola, S.; De Stefano, M.C.; Amaldi, F.; et al. Synthesis and function of ribosomal proteins--fading models and new perspectives. FEBS J. 2009, 276, 3199–3210.
- 92.
Fumagalli, S.; Di Cara, A.; Neb-Gulati, A.; et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 2009, 11, 501–508.
- 93.
Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; et al. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189.
- 94.
Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377.
- 95.
Llanos, S.; Clark, P.A.; Rowe, J.; et al. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452.
- 96.
Kurki, S.; Peltonen, K.; Latonen, L.; et al. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 2004, 5, 465–475.
- 97.
Colombo, E.; Marine, J.C.; Danovi, D.; et al. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533.
- 98.
Quin, J.; Chan, K.T.; Devlin, J.R.; et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 2016, 7, 49800–49818.
- 99.
Liu, T.; Pan, G.; Zhang, J.; et al. Molecular basis of CX-5461-induced DNA damage response in primary vascular smooth muscle cells. Heliyon 2024, 10, e37227.
- 100.
Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166.
- 101.
Panwar, V.; Singh, A.; Bhatt, M.; et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375.
- 102.
Goudarzi, K.M.; Nistér, M.; Lindström, M.S.; et al. mTOR inhibitors blunt the p53 response to nucleolar stress by regulating RPL11 and MDM2 levels. Cancer Biol. Ther. 2014, 15, 1499–1514.
- 103.
Nishimura, K.; Kumazawa, T.; Kuroda, T.; et al.Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. 2015, 10, 1310–1323.
- 104.
Yuan, X.; Zhou, Y.; Casanova, E.; et al. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell. 2005, 19, 77–87.
- 105.
Liao, H.; Gaur, A.; Mauvais, C.; et al. p53 induces a survival transcriptional response after nucleolar stress. Mol. Biol. Cell 2021, 32, ar3.
- 106.
Goff, S.L.; Boussaid, I.; Floquet, C.; et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood 2021, 137, 89–102.
- 107.
van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309.
- 108.
Wang, H.T.; Chen, T.Y.; Weng, C.W.; et al. Acrolein preferentially damages nucleolus eliciting ribosomal stress and apoptosis in human cancer cells. Oncotarget 2016, 7, 80450–80464.
- 109.
Mekhail, K.; Gunaratnam, L.; Bonicalzi, M.E.; et al. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat. Cell Biol. 2004, 6, 642–647.
- 110.
Guo, W.; Yang, Z.; Xia, Q.; et al. Arsenite stabilizes HIF-1α protein through p85α-mediated up-regulation of inducible Hsp70 protein expression. Cell Mol. Life Sci. 2011, 68, 475–488.
- 111.
Keil, M.; Meyer, M.T.; Dannheisig, D.P.; et al. Loss of Peter Pan protein is associated with cell cycle defects and apoptotic events. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 882–895.
- 112.
Pfister, A.S.; Keil, M.; Kühl, M. The Wnt Target Protein Peter Pan Defines a Novel p53-independent Nucleolar Stress-Response Pathway. J. Biol. Chem. 2015, 290, 10905–10918.
- 113.
Chen, J.; Stark, L.A. Insights into the relationship between nucleolar stress and the NF-kappaB pathway. Trends Genet. 2019, 35, 768–780.
- 114.
Chen, J.; Lobb, I.T.; Morin, P.; et al. Identification of a novel TIF-IA-NF-κB nucleolar stress response pathway. Nucleic Acids Res. 2018, 46, 6188–6205.
- 115.
Khandelwal, N.; Simpson, J.; Taylor, G.; et al. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ. 2011, 18, 1889–1903.
- 116.
Lessard, F.; Brakier-Gingras, L.; Ferbeyre, G. Ribosomal proteins control tumor suppressor pathways in response to nucleolar stress. Bioessays 2019, 41, e1800183.
- 117.
Tiku, V.; Antebi, A. Nucleolar function in lifespan regulation. Trends Cell Biol. 2018, 28, 662–672.
- 118.
Bursać, S.; Prodan, Y.; Pullen, N.; et al. Dysregulated ribosome biogenesis reveals therapeutic liabilities in cancer. Trends Cancer 2021, 7, 57–76.
- 119.
Orgebin, E.; Lamoureux, F.; Isidor, B.; et al. Ribosomopathies: New therapeutic perspectives. Cells 2020, 9, 2080.
- 120.
Yan, D.; Hua, L. Nucleolar stress: Friend or foe in cardiac function? Front. Cardiovasc. Med. 2022, 9, 1045455.
- 121.
Unverferth, B.J.; Magorien, R.D.; Balcerzak, S.P.; et al. Early changes in human myocardial nuclei after doxorubicin. Cancer. 1983, 52, 215–221.
- 122.
Lambertenghi-Deliliers, G.; Zanon, P.L.; Pozzoli, E.F.; et al. Myocardial injury induced by a single dose of adriamycin: An electron microscopic study. Tumori 1976, 62, 517–528.
- 123.
Leblanc, B.; Mompon, P.R.; Espérandieu, O.; et al. Nucleolar organizer regions in cardiac lesions induced by doxorubicin. Toxicol. Pathol. 1991, 19, 176–183.
- 124.
Roselló-Lletí, E.; Rivera, M.; Cortés, R.; et al. Influence of heart failure on nucleolar organization and protein expression inhuman hearts. Biochem. Biophys. Res. Commun. 2012, 418, 222–228.
- 125.
Tsai, R.Y.L. Turning a new page on nucleostemin and self-renewal. J. Cell Sci. 2014, 127 Pt 18, 3885–3891.
- 126.
Scott, D.D.; Oeffinger, M. Nucleolin and nucleophosmin: Nucleolar proteins with multiple functions in DNA repair. Biochem. Cell Biol. 2016, 94, 419–432.
- 127.
Haaf, T.; Ward, D.C. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp. Cell Res. 1996, 224, 163–173.
- 128.
Siddiqi, S.; Gude, N.; Hosoda, T.; et al. Myocardial induction of nucleostemin in response to postnatal growth and pathological challenge. Circ. Res. 2008, 103, 89–97.
- 129.
Tang, Y.; Lin, X.; Chen, C.; et al. Nucleolin improves heart function during recovery from myocardial infarction by modulating macrophage polarization. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 386–395.
- 130.
Colombo, E.; Alcalay, M.; Pelicci, P.G. Nucleophosmin and its complex network: A possible therapeutic target in hematological diseases. Oncogene 2011, 30, 2595–2609.
- 131.
Frehlick, L.J.; Eirín-López, J.M.; Ausió, J. New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. Bioessays 2007, 29, 49–59.
- 132.
Colombo, E.; Bonetti, P.; Denchi, E.L.; et al. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol. Cell Biol. 2005, 25, 8874–8886.
- 133.
Csiszar, A.; Wang, M.; Lakatta, E.G.; et al. Inflammation and endothelial dysfunction during aging: Role of NF-kappaB. J. Appl. Physiol. 2008, 105, 1333–1341.
- 134.
Beji, S.; D’agostino, M.; Gambini, E.; et al. Doxorubicin induces an alarmin-like TLR4-dependent autocrine/paracrine action of Nucleophosmin in human cardiac mesenchymal progenitor cells. BMC Biol. 2021, 19, 124.
- 135.
Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173.
- 136.
Kinumi, T.; Ogawa, Y.; Kimata, J.; et al. Proteomic characterization of oxidative dysfunction in human umbilical vein endothelial cells (HUVEC) induced by exposure to oxidized LDL. Free Radic. Res. 2005, 39, 1335–1344.
- 137.
Kim, Y.; Nurakhayev, S.; Nurkesh, A.; et al. Macrophage polarization in cardiac tissue repair following myocardial infarction. Int. J. Mol. Sci. 2021, 22, 2715.
- 138.
Gudkova, A.; Shliakhto, E.V.; Mamaev, N.N.; et al. Elevated expression of argentophilic proteins from the nucleolar organizer regions in myocardium of patients with obstructive hypertrophic cardiomyopathy and mutations in p53 tumor suppressor gene. Tsitologiia 2003, 45, 1124–1133.
- 139.
Monte, E.; Mouillesseaux, K.; Chen, H.; et al. Systems proteomics of cardiac chromatin identifies nucleolin as a regulator of growth and cellular plasticity in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1624–H1638.
- 140.
Lindstrom, M.S.; Jurada, D.; Bursac, S.; et al. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366.
- 141.
Tsekrekou, M.; Stratigi, K.; Chatzinikolaou, G. The nucleolus: In genome maintenance and repair. Int. J. Mol. Sci. 2017, 18, 1411.
- 142.
Mamaev, N.N.; Gudkova, A.Y.; Amineva, K.K. AgNORs in the myocardium in ischaemic heart disease complicated by heart failure: A postmortem study. Mol. Pathol. 1998, 51, 102–104.
- 143.
Trere, D. AgNOR staining and quantification. Micron 2000, 31, 127–131.
- 144.
Mamaev, N.N.; Kovalyeva, O.V.; Amineva, K.K.; et al. AgNORs in cardiomyocytes from surgical patients with coronary heart disease. Mol. Pathol. 1998, 51, 218–221.
- 145.
Gudkova, A.; Amineva, K.K.; Mamaev, N.N. Activity of the nucleolar organizers in cardiomyocytes of patients with arterial hypertension of varying genesis. Arkh Patol. 1989, 51, 55–58.
- 146.
Lee, C.; Smith, B.A.; Bandyopadhyay, K.; et al. DNA damage disrupts the p14ARF-B23 (nucleophosmin) interaction and triggers a transient subnuclear redistribution of p14ARF. Cancer Res. 2005, 65, 9834–9842.
- 147.
Ruan, K.; Bai, G.; Fang, Y.; et al. Biomolecular condensates and disease pathogenesis. Sci. China Life Sci. 2024, 67, 1792–1832.
- 148.
Horii, Y.; Matsuda, S.; Toyota, C.; et al. VGLL3 is a mechanosensitive protein that promotes cardiac fibrosis through liquid-liquid phase separation. Nat. Commun. 2023, 14, 550.
- 149.
Xie, S.; Chen, M.; Fang, W.; et al. Diminished arachidonate 5-lipoxygenase perturbs phase separation and transcriptional response of Runx2 to reverse pathological ventricular remodeling. EBioMedicine 2022, 86, 104359.
- 150.
Jiang, Y.; Gu, J.; Niu, X.; et al. Phosphorylation-Regulated Dynamic Phase Separation of HIP-55 Protects Against Heart Failure. Circulation 2024, 150, 938–951.
- 151.
Schneider, J.W.; Oommen, S.; Qureshi, M.Y.; et al. Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat. Med. 2020, 26, 1788–1800.
- 152.
Scholz, D.; Baicu, C.F.; Tuxworth, W.J.; et al. Microtubule-dependent distribution of mRNA in adult cardiocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1135–H1144.
- 153.
Li, X.; Zhu, R.; Jiang, H.; et al. Autophagy enhanced by curcumin ameliorates inflammation in atherogenesis via the TFEB-P300-BRD4 axis. Acta Pharm. Sin. B 2022, 12, 2280–2299.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com