G protein-coupled receptors (GPCRs) are key mediators of cellular signaling, governing fundamental physiological and pathophysiological processes. This central role establishes them as prominent drug targets for a wide range of diseases. The concept of GPCR biased signaling was initially defined by the receptor’s ability to differentially engage G proteins versus β-arrestins. Research has since broadened this paradigm to reveal diverse mechanisms, including preferential coupling to specific Gα subtypes, spatially segregated signaling, regulation by post-translational modifications (e.g., phosphorylation), and distinct outputs from receptor oligomers. Together, these findings illuminate the complex signaling repertoire of GPCRs. Leveraging biased signaling to activate beneficial pathways, therefore offers a compelling path toward therapeutics with enhanced efficacy and reduced adverse effects. This review explores the evolution of GPCR biased signaling concepts and evaluates the current pipeline of investigational and approved drugs emerging from this paradigm.
- Open Access
- Review
GPCR Biased Signaling: Conceptual Advancements and Therapeutic Innovation
- Supeng Li 1,2,†,
- Yongyin Huang 1,†,
- Xingsheng Sun 2,*,
- Ao Shen 1,*
Author Information
Received: 21 Jul 2025 | Revised: 30 Sep 2025 | Accepted: 12 Oct 2025 | Published: 09 Jun 2026
Abstract
Keywords
G protein-coupled receptors | β-arrestin signaling | spatial bias | post-translational modification bias | oligomerization bias
1. Introduction
G protein-coupled receptors (GPCRs), also known as 7-transmembrane receptors (7TMRs), constitute the largest and most diverse superfamily of human proteins. Encompassing over 800 members, they orchestrate a vast array of cellular and physiological processes. GPCRs detect extracellular stimuli—such as light, ions, hormones, and neurotransmitters—and transduce these signals via intracellular second messengers, including cAMP and cGMP [1]. Early models posited that GPCR signaling occurred exclusively through G protein-dependent pathways. However, the discovery of β-arrestin-mediated signaling fundamentally upended this paradigm. Subsequent findings revealed that certain ligands selectively activate either G protein or β-arrestin pathways at specific GPCRs, resulting in distinct functional outcomes [2]. This discovery gave rise to the concept of “biased signaling”, defined as the ligand-directed activation of divergent signaling cascades that elicit distinct biological responses [3].
Recent advances have uncovered a spectrum of novel bias models, while classical concepts of biased signaling based on G protein or β-arrestin selectivity is now recognized as limited [4,5]. These include Gα subtype bias (selective activation of specific Gα protein subtypes), spatial bias (compartmentalized signaling in distinct subcellular locations), receptor post-translational modification bias, and oligomerization bias (signaling based on receptor assembly states). This expanded understanding of GPCR signaling has profound potential implications for the development of novel therapeutics in precision medicine [6]. Notably, although approximately one-third of clinically used drugs act by targeting GPCRs, the majority of them lack “biased signaling” characteristics. This review briefly summarizes the latest conceptual advancements in GPCR biased signaling. By harnessing the ligand selectivity toward these biased signaling cascades, GPCR pharmacology holds promise for developing more precise therapies that maximize efficacy while minimizing side effects.
2. β-Arrestin Biased Signaling
GPCRs share a conserved structural architecture: seven transmembrane helices (TM1–TM7) are connected by three extracellular loops (ECLs) and three intracellular loops (ICLs), and terminate in an intracellular C-terminal tail [7]. Upon agonist binding, GPCRs typically recruit G protein-coupled receptor kinases (GRKs), which phosphorylate specific serine and threonine residues primarily on the receptor’s C-terminal tail or third intracellular loop (ICL3) [8]. This phosphorylation creates high-affinity binding sites for β-arrestins. The recruited β-arrestins not only sterically hinder G protein coupling, thereby inducing receptor desensitization, but also initiate receptor internalization [9]. Consequently, β-arrestins (β-arrestin-1 and β-arrestin-2) were initially characterized as mediators of GPCR desensitization [10]. However, they are now recognized as bona fide adaptor proteins that relay signals to diverse downstream effectors, supporting more complex regulatory functions. Notably, G protein pathways and the β-arrestin-mediated signaling pathways are often spatially and temporally segregated, allowing them to mediate distinct physiological and pathophysiological outcomes [11] (Figure 1A). The angiotensin II type 1 receptor (AT1R) exemplifies this concept. In AT1R signaling, G protein signaling drives potent vasoconstriction and elevates blood pressure [12]. Conversely, β-arrestin-biased signaling can elicit potentially beneficial responses, such as anti-apoptotic effects [13]. Therefore, developing biased ligands for AT1R, which are capable of selectively engaging either G protein- or β-arrestin-mediated signaling, holds therapeutic promise by preferentially promoting beneficial responses while mitigating detrimental effects tied to receptor activation [14]. A key example is the endogenous heptapeptide angiotensin-(1-7) (Ang-1-7), a naturally occurring β-arrestin-biased ligand for AT1R. Unlike the canonical agonist Ang II, Ang-1-7 binds AT1R without activating G protein-mediated pathways linked to hypertension and cardiac hypertrophy. Instead, it selectively triggers cardioprotective β-arrestin signaling [15,16].
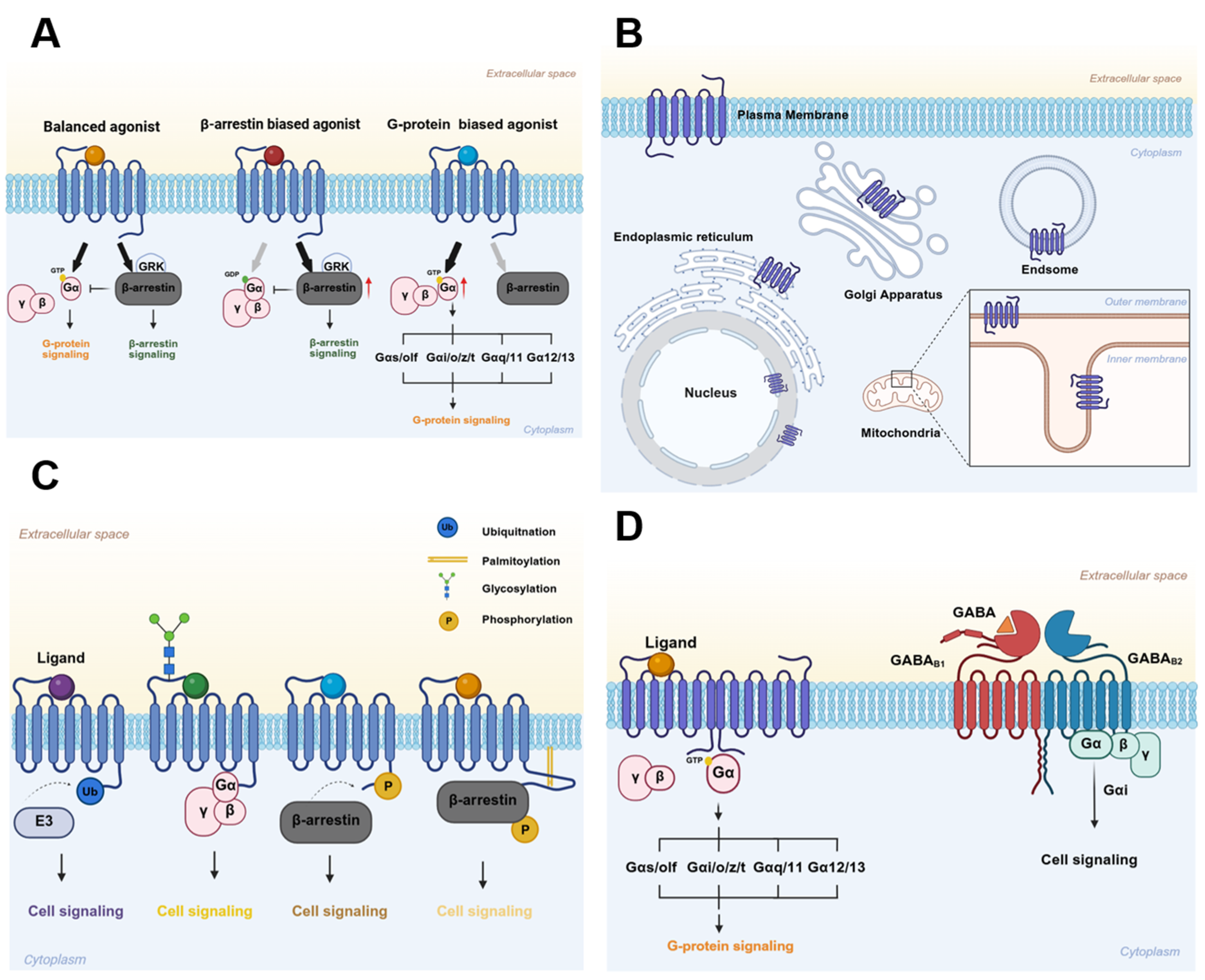
Emerging evidence now challenges the classical paradigm of mutually exclusive G protein and β-arrestin signaling, revealing greater functional diversity for β-arrestin. A notable example comes from the vasopressin type 2 receptor (V2R), which can form stable “super-complexes” (or “megaplexes”) comprising a single receptor simultaneously bound to both a G protein and a β-arrestin [17]. Cryo-EM structural analyses and functional studies show that within these complexes, the G protein engages the receptor’s intracellular core, while β-arrestin binds the phosphorylated C-terminal tail, with all three components maintaining their canonically active conformations [18]. These super-complexes specifically facilitate sustained G protein signaling from endosomal compartments, thereby enabling prolonged cellular responses. As the progress in β-arrestin-biased signaling has been thoroughly summarized in several recent reviews [19,20,21,22], we will not elaborate on this aspect in the present discussion.
3. Gα Subtype Biased Signaling
Heterotrimeric G protein complexes, which consist of Gα, Gβ, and Gγ subunits, are categorized into major families according to their sequence homology and downstream effector specificity. These include the Gαs/olf, Gαi/o/z/t, Gαq/11, and Gα12/13 subfamilies. Gαs stimulates adenylyl cyclase (AC) to elevate cAMP levels and activate protein kinase A (PKA) signaling, whereas Gαi inhibits AC, establishing bidirectional control over this pathway [19]. In retinal cells, Gαt specifically mediates phototransduction [23,24]. When a single GPCR activates multiple Gα subtypes, its preferential coupling to specific G proteins forms the basis of Gα subtype-biased signaling [25,26] (Figure 1A). Although structural studies attribute this selectivity to the conformational properties of the Gα C-termini [27], the mechanisms that dynamically regulate Gα subtype preference among homologous receptors remain incompletely understood.
In the heart, both β1- and β2-adrenergic receptors (β1AR/β2AR) exhibit dual coupling to Gαs and Gαi, resulting in divergent signaling outcomes. β1AR was originally thought to signal exclusively through Gαs, which stimulates AC to increase cAMP. Recent evidence, however, shows that it also couples to Gαi [28,29,30], an interaction implicated in cardioprotection via cGMP pathways [31]. Meanwhile, β2AR canonically activates Gαs to raise cAMP in cardiomyocytes, but it also recruits Gαi. This Gαi coupling not only inhibits AC and counterbalances Gαs signaling [32], but also activates additional effectors such as mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) or phosphatidylinositol 3′-kinase (PI3K) [33]. The two pathways also display distinct temporal profiles: Gαs-mediated cAMP responses are typically rapid and transient, while Gαi-dependent MAPK activation develops more slowly but is sustained [34]. This Gαi coupling is a less direct process [35] due to requirements for receptor phosphorylation [36] and β-arrestin recruitment [37] prior to G protein binding. Notably, β2AR’s preference for Gα subtypes may be influenced by local plasma membrane charge [38], which is modulated by phospholipids and ions such as Ca2⁺.
Beyond βARs, the muscarinic acetylcholine receptor type 3 (M3R) exemplifies another GPCR capable of engaging multiple Gα proteins [39]. Historically, M3R was known to couple to the Gαq signaling pathway to regulate vascular tone, bronchoconstriction, and insulin secretion [39]; recent ligand-binding and GTP turnover assays have demonstrated that it can also couple productively to Gαi and Gαs [40]. This dual coupling activates canonical second messenger cascades—including cAMP and inositol trisphosphate (IP₃) signaling—establishing broader G protein promiscuity than previously recognized and expanding M3R’s complex regulatory network.
4. Spatial Bias in GPCRs Signaling: Mechanisms and Therapeutic Potential
Traditionally, GPCR signaling was thought to operate mostly at the plasma membrane. But we now know GPCRs act in many subcellular compartments—including the endosomes [41], mitochondria [42], endoplasmic reticulum [43], Golgi apparatus [44], and nuclear envelope [45] (Figure 1B). Finding these intracellular GPCRs has opened up a new way to understand how GPCR signaling actually works [46,47]. The activation of compartmentalized GPCRs generates spatially segregated signaling microdomains [45,48], enabling cells to elicit divergent physiological responses. These specialized spots fine-tune cell functions in two ways: they recruit unique effector molecules, or they activate common ones but with different timing. This spatial control is exactly why they’re such promising targets for precisely tweaking GPCR signaling.
4.1. Plasma Membrane (PM)
Plasma membrane (PM) microdomains create tiny, localized signaling hubs where cAMP levels are naturally higher than in the bulk cytosol [49]. This sharp cAMP gradient even lets low-dose agonists preferentially activate PKA that’s stuck to the membrane [50]. The limited diffusion of cAMP into the cytosol, resulting from saturation of cytosolic buffering proteins [51], further reinforces these compartmentalized signaling gradients [52,53]. In previous work, we showed that GRK- and PKA-mediated phosphorylation dynamically shifts β2AR populations between the PM and endosomes [54]. Interestingly, hydrophobic ligands such as carvedilol and alprenolol induce the formation of distinct, non-phosphorylated β2AR clusters. These clusters selectively amp up Gαs signaling, resulting in the assembly of cAMP/PKA nanodomains that phosphorylate L-type calcium channels with pinpoint spatiotemporal accuracy [55]. This nanoscale organization thus enables precise functional specification even among seemingly homogeneous receptor populations.
4.2. Endosomes
Endosomes represent a major signaling platform that extends GPCR activity beyond the plasma membrane. Numerous GPCRs continue to activate G proteins from endosomal compartments, producing signaling responses that differ temporally and spatially from those initiated at the plasma membrane [56]. A well-studied example is the parathyroid hormone receptor (PTHR), which signals from both locales [57]. When activated at the plasma membrane, PTHR elicits transient cAMP pulses that mediate acute metabolic regulation. In contrast, endosomal PTHR generates sustained cAMP waves that ultimately govern transcriptional programs [58,59].
Similarly, studies employing conformation-specific nanobodies demonstrated that the β2AR activates Gαs not only at the plasma membrane but also subsequently from early endosomes, generating two distinct phases of cAMP production [41]. This endosomal cAMP pool was revealed to be essential for cAMP-dependent transcription, underscoring that the location of cAMP production directly determines functional outcomes [60]. Interestingly, research on receptors such as vasoactive intestinal peptide receptor 1 (VIPR1) and V2R has demonstrated that some GPCRs can internalize and activate Gαs in endosomes independently of β-arrestin and β-arrestin plays a key role in sculpting the spatiotemporal profile of cellular GPCR–G protein signaling through location-specific remodeling of GPCR-β-arrestin complexes [61,62].
The physiological relevance of endosomal GPCR signaling is underscored by its critical roles in processes such as calcium homeostasis and pain perception. This has prompted the exploration of therapeutic strategies that precisely target the endosomal GPCR pool, including the design of pH-sensitive nanoparticles and cholestanol-conjugated ligands, to achieve spatially biased therapeutic effects [56].
4.3. Sarcoplasmic Reticulum (SR)
Cardiomyocytes have a specialized compartment—the sarcoplasmic reticulum (SR)—that acts as a command center for localized cAMP signaling, a linchpin of heart function [63,64,65,66]. These precise signals stem from a unique pool of β1ARs nestled on the SR membrane, which generate tight, localized cAMP bursts to fine-tune excitation-contraction coupling—whether the heart is healthy or failing [67]. The pathway operates through a dedicated substrate cycle: norepinephrine (NE) enters the cardiomyocyte via the organic cation transporter 3 (OCT3) [68] to activate these SR-localized β1Ars [65]. This activation produces a localized cAMP domain that modulates the activity of the SR calcium pump (SERCA) through PKA-dependent phosphorylation [64]. Monoamine oxidase A (MAOA) keeps this signaling in check: it breaks down cytosolic NE [66]. In heart failure, MAOA gets overexpressed—dimming the SR-β1AR signal and sapping the heart’s contractile strength. But inhibiting MAOA enhances cardiomyocyte contractility and restores systolic function, underscoring the therapeutic potential of modulating this specific axis [67]. In the context of heart failure, MAOA upregulation leads to diminished SR-β1AR signaling and impaired contractility. Conversely, inhibiting MAOA enhances cardiomyocyte contractility and restores systolic function, underscoring the therapeutic potential of modulating this specific axis [65]. Together, OCT3 and MAOA constitute an upstream regulatory unit that governs the spatial specificity and functional output of SR-β1AR signaling.
4.4. Golgi Apparatus
The functional segregation of cAMP signaling between the Golgi apparatus and plasma membrane represents a critical regulatory mechanism in cardiac physiology [69]. This spatial specificity is dictated by ligand-specific trafficking, wherein the human β1AR can initiate non-canonical Gαs signaling from pre-existing Golgi pools, a process controlled by the physicochemical properties of the ligand [44]. Functionally, Golgi-generated cAMP selectively phosphorylates phospholamban (PLB) to accelerate cardiac relaxation, whereas PM-derived cAMP primarily enhances contractility through ryanodine receptor (RyR2) phosphorylation [70]. Compartmental access of ligands follows a defined lipophilicity hierarchy: adrenaline requires OCT3-dependent transport to activate the Golgi-PLB pathway, whereas highly lipophilic agonists such as dobutamine diffuse directly across membranes to engage Golgi-resident β1ARs. This principle extends to clinically relevant β-blockers: hydrophobic antagonists including metoprolol inhibit β1AR signaling at both PM and Golgi membranes, whereas hydrophilic agents like sotalol selectively block PM-localized signaling without affecting the Golgi pool [44]. Thus, lipophilicity-dependent ligand delivery governs signaling specificity by determining the subcellular site of receptor activation. This mechanism establishes a molecular basis for targeting compartment-specific cAMP pathways, offering a rationale for developing precision therapies for diastolic dysfunction.
5. PTM-Based Biased Signaling in GPCRs
Post-translational modifications (PTMs) such as phosphorylation, ubiquitination, and glycosylation serve as critical regulators of GPCR function [71] (Figure 1C). Post-translational modifications (PTMs) such as phosphorylation, ubiquitination, and glycosylation serve as critical regulators of GPCR function [72]. A prime example is found in the regulation of the β-adrenergic receptors (βARs). Phosphorylation of specific C-terminal residues (S355/S356) by GRK2 enhances β-arrestin binding, which in turn robustly promotes clathrin-mediated endocytosis [73]; conversely, impaired GRK2-mediated phosphorylation prevents internalization [74]. In contrast, PKA phosphorylates distinct sites on the third intracellular loop (S261/S262) and C-terminus (S345/S346), promoting receptor dimerization and plasma membrane retention [54,75]. This site-specific phosphorylation creates a spatial “barcode” wherein the pattern of modification, rather than its overall level, dictates β-arrestin recruitment [76]. Our recent work demonstrated that these distinct PTMs segregate β2AR into two spatially separate populations within a single cell: GRK-phosphorylated monomers undergo internalization, while PKA-phosphorylated dimers remain membrane-bound [54]. This PTM-specific partitioning represents a novel mechanism for functional bias.
Genetic variation in the human β1AR profoundly influence its post-translational regulation and downstream signaling [69]. Two common single-nucleotide polymorphisms (SNPs) highlight this: the S49G substitution in the extracellular N-terminus and the R389G substitution in the intracellular C-terminus [77]. These variants confer distinct functional properties: the C-terminal R389 variant exhibits increased norepinephrine-induced phosphorylation, leading to enhanced adenylyl cyclase (AC) activation, cAMP production, and β-arrestin recruitment compared to the G389 variant [78]. At the N-terminus, the G49 variant shows higher basal and agonist-stimulated AC activity than the S49 variant, with altered desensitization kinetics [79]. This functional divergence appears driven by O-linked glycosylation, which preferentially modifies the extracellular domain of the G49 variant and helps define its unique PTM signature [80]. Intriguingly, the S49 variant promotes increased isoproterenol-mediated β-arrestin recruitment, an effect that may functionally compensate for the signaling intensity associated with the C-terminal R389G polymorphism [81].
6. Receptor Oligomerization-Dependent Signaling Bias
Many GPCRs exist as monomers or homo-/hetero-oligomers—a structural flexibility that dictates which distinct signaling pathways they activate (Figure 1D). Oligomerization induces specific conformational rearrangements that tweak receptor trafficking and effector coupling, allowing a single receptor type to trigger divergent cellular responses even when bound to the same ligand. This oligomerization-dependent signaling bias is now regarded as a core mechanism shaping GPCR functional diversity.
Class C GPCRs canonically require dimerization for function. For example, metabotropic glutamate (mGlu) receptors activate G proteins (Gαq, Gα11, or Gαi/o) only as homodimers, via rearrangements in their transmembrane domains, whereas their monomeric forms are inactive [82,83]. Similarly, the GABAB receptor mandates heterodimerization between the GABAB1 (ligand-binding) and GABAB2 (G protein-coupling) subunits to enable Gαi-mediated signaling [82,83].
Although Class A GPCRs were once thought to act as lone monomers, growing evidence reveals that homo- and heterodimerization reshapes their pharmacological behavior in profound ways. For instance, the platelet-activating factor receptor (PAFR) dimerizes via TM 1 and TM 4/5. Chemically stabilizing this interface enhances Gαq signaling while attenuating β-arrestin recruitment and receptor internalization [84]. A natural genetic variant, PAFR E178K, promotes dimer formation and similarly augments Gαq efficacy while reducing internalization [84].
Heterodimerization adds another layer of complexity, creating hybrid receptors with new pathway biases and functional cross-talk. A clear illustration is provided by the 5-hydroxytryptamine 2A receptor (5-HT2A), which primarily activates Gαq/11-phospholipase C (PLC)-calcium pathways as a monomer. Upon heterodimerization with mGluR2, however, its signaling output shifts towards Gαi/o pathways, enabling dual Gαq/11 and Gαi/o activation [85]. This paradigm holds significant physiological and pathological relevance. For instance, PAFR oligomerization contributes to inflammatory pathologies [86,87], and 5-HT2AR-mGluR2 heterodimers are linked to psychotic disorders [88,89]. Similarly, β2AR-5-HT2BR heterodimers shift signaling from Gαs to Gαi, a mechanism associated with cardioprotective effects [90]. Conversely, heterodimerization between the apelin receptor (APJ) and bradykinin B1R suppresses Gαi signaling while enhancing Gαq activation [91]. The functional impact can also extend to signaling kinetics. While μ-opioid receptor (μOR) homodimers mediate only transient, β-arrestin-dependent ERK1/2 phosphorylation, the μOR-δOR heterodimer sustains this phosphorylation [92]. Furthermore, CXCR4-CXCR7 heterodimerization reduces canonical CXCR4-mediated Gαi signaling, while simultaneously potentiating β-arrestin-mediated activation of the ERK1/2 and MAPK cascades [93]. These collective findings establish GPCR heterodimerization as a fundamental mechanism for generating signaling diversity and bias.
7. Biased Therapeutics in Cardiovascular and Neural Systems
While the conceptual framework for GPCR biased signaling has expanded to include novel mechanisms beyond the classical G protein/β-arrestin paradigm, translational drug development remains overwhelmingly focused on β-arrestin-biased ligands. This focus is largely driven by the pathway’s currently superior translational validation, which has, however, overshadowed the therapeutic potential of other emerging modes of bias.
7.1. β-Adrenergic Receptors (βARs)
βARs are critical regulators of cardiac contractility and heart rate. The three subtypes (β1, β2, and β3) are differentially expressed, with β1AR and β2AR being the predominant isoforms in the heart. Although acute βAR stimulation is a powerful mechanism for increasing cardiac output during ‘fight-or-flight’ responses, chronic β1AR activation promotes cardiomyocyte hypertrophy and apoptosis [94,95,96] and is a hallmark of heart failure development and progression [97,98]. Consequently, β-blockers (particularly β1-selective antagonists) are mainstays in the clinical management of cardiovascular diseases, including hypertension [99], arrhythmias [100], and chronic heart failure [101]. Among these agents, carvedilol was long thought to act as a β-arrestin-biased agonist at the β1AR [31], a property once believed to underlie its superior clinical efficacy compared to neutral antagonists [102]. However, recent studies have revealed that carvedilol instead selectively enhances a β1AR-mediated cGMP signal via a Gαi-NOS axis [103]. In contrast to the cardiotoxic effects of chronic β1AR activation, β2AR signaling is generally cardioprotective, activating anti-apoptotic and anti-necrotic pathways [20,104,105]. Carvedilol thus exhibits a fundamental mechanistic divergence between β1AR and β2AR signaling. At β2AR, its cardioprotective effects depend exclusively on the Gαs/cAMP pathway [106,107]. At the β1AR, by contrast, it concurrently antagonizes cardiotoxic Gαs signaling [108] and activates the protective Gαi-eNOS-cGMP axis [103]. Furthermore, fenoterol has been identified as a Gαs-biased agonist of β2ARs and has been shown to restore myocardial contractility [109,110].
7.2. Angiotensin II Type 1 Receptor (AT1R)
AT1R is widely expressed within the cardiovascular system and is a critical regulator of cardiac pathophysiology. Pathological AT1R activation promotes myocardial hypertrophy and fibrosis [111], establishing it as a common target for blockers used to treat cardiovascular diseases [112]. As AT1R signals through both Gαq and β-arrestin pathways, it represents a pivotal system for studying signaling bias. For instance, a single amino acid mutation in angiotensin II itself can induce robust β-arrestin-biased signaling at AT1R [113]. This mutated Ang II (SII) binds to AT1R, recruits β-arrestin, and promotes receptor internalization without activating G protein coupling [114,115]. By leveraging this principle, several β-arrestin-biased AT1R antagonists (e.g., TRV120023, TRV120026, TRV120027) have been developed, showing promising efficacy in preclinical models [116,117,118].
7.3. Adenosine A1 Receptor (A1AR)
The adenosine A1 receptor (A1AR) is ubiquitously expressed in cardiomyocytes and vascular smooth muscle cells. Upon activation, A1ARs initiate Gαi-mediated signaling, which confers robust cardio protection against ischemia-reperfusion injury [119]. However, traditional unbiased agonists such as N6-cyclohexyladenosine simultaneously activate detrimental Ca2⁺ influx pathways, leading to clinically limiting bradycardia and hypotension. To overcome these limitations, the biased agonist VCP746 was developed to selectively activate the beneficial Gαi/cAMP pathway while sparing Ca2⁺-related adverse effects [120]. In preclinical validation, VCP746 effectively protected rat cardiomyocytes from ischemic injury without affecting atrial heart rate [121]. This biased profile was further explored with the orally active agonist neladenoson, which was specifically designed to avoid Ca2⁺ and MAPK pathway activation. While neladenoson worked to prevent bradycardia in healthy volunteers, it ultimately flopped in a Phase II trial for chronic heart failure. This clinical miss highlights the ongoing challenge of optimizing pathway selectivity for effective chronic disease management [122,123].
7.4. The Apelin Receptor (APJ)
The apelin receptor (APJ), activated by its endogenous peptide apelin, serves as a central hub in maintaining cardiovascular homeostasis and represents a promising therapeutic target for cardiovascular diseases [124]. APJ signaling operates via two principal pathways—G protein axis and β-arrestin axis—that mediate distinct functional outcomes. The endogenous ligand apelin-13 engages both Gαi and β-arrestin arms: Gαi signaling confers cardioprotection by boosting eNOS activity and suppressing myocardial apoptosis, whereas β-arrestin activation drives pathological myocardial hypertrophy via the ERK1/2 cascade. These opposing effects have hampered the translational potential of unbiased APJ agonists. To uncouple beneficial signaling from deleterious effects, several Gαi-biased APJ agonists have been rationally designed. An early example, MM07, demonstrated enhancing cardiac output more effectively than apelin-13 [125] and induced twice the magnitude of forearm blood flow dilation in human volunteers. Subsequent efforts yielded Bpa91, a Gαi-biased analog engineered by substituting the C-terminal phenylalanine of apelin-13 with p-benzoyl-l-phenylalanine [126]. More recently, structural studies pinpointed “twin hotspots” within APJ as critical determinants of signaling bias. Building on this insight, the structure-guided design led to WN561—a highly selective G protein–biased agonist that exhibits superior anti-hypertrophic efficacy and a refined safety profile compared to conventional APJ agonists [127].
7.5. Opioid Receptors
Opioid receptors (μOR, κOR, δOR) are GPCRs that transduce the effects of opioids, playing critical roles in pain, reward, and addiction [128,129]. While classic opioids like morphine exert analgesia primarily through μOR-mediated G protein signaling, many severe side effects (e.g., respiratory depression, addiction) stem from μOR-β-arrestin pathways [129,130,131]. This understanding has driven the development of G protein-biased μOR ligands, such as PZM21 [132] and SR-17018 [133], that offer effective analgesia in animal models and significantly mitigate adverse effects like respiratory depression and tolerance. For κOR agonists, which avoid respiratory depression but can cause dysphoria and sedation, studies reveal that β-arrestin2 signaling underlies these dysphoric effects [134]. Thus, developing G protein-biased ligands that reduce β-arrestin engagement represents a promising strategy for advancing safer opioid therapeutics.
7.6. Dopamine Receptors (DRs)
DRs, highly expressed in the brain, fall into two families: D1-like (D1, D5) and D2-like (D2, D3, D4) [135]. The D2 receptor (D2R) serves as a key model for developing precise neuropsychiatric therapeutics. For instance, β-arrestin-biased D2R ligands like UNC9975 and UNC9994 selectively engage β-arrestin over G protein pathways, ameliorating schizophrenia-like behaviors in animals with fewer side effects than conventional antipsychotics [136], highlighting the therapeutic potential of D2R bias. Conversely, bias at the D1 receptor (D1R) involves differential activation of the highly homologous Gαs and Gαolf proteins. Ligands such as dihydrexidine (DHX) act as full agonists for Gαs but partial agonists for Gαolf. Structural studies reveal that a salt bridge between Gαs Arg38 and D1R Glu132, which is disrupted in Gαolf, underlies this bias [137]. Furthermore, the regional expression of Gαs (predominantly in the cortex) versus Gαolf (enriched in the striatum) confers brain region-specific activity to D1R ligands, adding a critical layer of functional selectivity.
7.7. 5-Hydroxytryptamine 2A Receptor (5-HT2AR)
The 5-HT2A receptor, a primary target of lysergic acid diethylamide (LSD) and related psychedelics, has emerged as a promising platform for disentangling biased signaling to separate antidepressant efficacy from hallucinogenic risk. Serotonin (5-hydroxytryptamine, 5-HT) is an evolutionarily conserved monoamine neurotransmitter that regulates key CNS functions, including mood, cognition, and pain perception. Activation of 5-HT2AR engages both Gαq signaling—associated with psychedelic effects—and β-arrestin pathways [138], disentangling which contribute to its antidepressant-like properties.
Structural studies of 5-HT2AR bound to serotonin, LSD, and the non-hallucinogenic agonist lisuride have identified specific ligand-receptor interactions that enhance β-arrestin recruitment. These insights have enabled the rational design of β-arrestin-biased agonists that elicit antidepressant effects in animal models without inducing hallucinations [139]. Further supporting this dissociation, a drug’s capacity to activate 5-HT2AR–Gαq signaling predicts its psychedelic potential, where a defined threshold of Gαq activation is required to elicit hallucinogenic effects. This mechanistic understanding explains why partial agonists such as lisuride, which do not reach this activation threshold at standard doses, lack hallucinogenic properties [140].
8. Conclusions and Perspectives
Understanding GPCR biased signaling is fundamental to explaining cellular complexity and creating novel precision therapeutics [141]. Evidence now reveals a dynamic and multi-faceted system where receptor activity is precisely tuned through distinct biased mechanisms—from β-arrestin recruitment and Gα subtype selectivity to subcellular localization and post-translational modifications [142,143]. Together, these mechanisms afford cells precise spatiotemporal control over GPCR signaling.
Leveraging this signaling bias for therapeutic benefit holds significant promise in developing safer, more effective treatments. But translating this potential into clinical reality remains tough—biased signaling’s journey from bench to bedside is still in its early days. Tackling this hurdle demands cutting-edge tools: FRET biosensors, nanobodies, super-resolution microscopy, and AI (e.g., AlphaFold)—all poised to probe bias at the molecular level. Meanwhile, grounding biased signaling in the pathological context of human disease is key to designing clinical interventions that hit the mark.
To summarize, the field of GPCR biased signaling is shifting from characterizing phenomenological drug-receptor interactions to achieving targeted pathway modulation. The ultimate goal is to selectively modulate GPCRs: activating the right signal, in the right cellular compartment, at the right time. Attaining this goal will undoubtedly transform therapeutic approaches for a broad spectrum of neurological, cardiovascular, and other diseases.
Author Contributions
S.L.: conceptualization, methodology, investigation, writing—original draft preparation, visualization. Y.H.: data curation, validation, writing—original draft preparation, software. X.S.: conceptualization, supervision, project administration, writing—review and editing. A.S.: supervision, funding acquisition, project administration, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful for financial support from the National Natural Science Foundation of China (82070406), and the Department of Science and Technology of Guangdong (2024A1515010574 and 2025A1515011882).
Conflicts of Interest
The authors declare no conflict of interest.
Use of AI and AI-Assisted Technologies
During the preparation of this work, the authors used ChatGPT for language refinement and proofreading. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
References
- 1.
Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. https://doi.org/10.1146/annurev-biochem-060614-033910.
- 2.
Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. https://doi.org/10.1124/pr.116.013367.
- 3.
Violin, J.D.; Lefkowitz, R.J. β-Arrestin-Biased Ligands at Seven-Transmembrane Receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. https://doi.org/10.1016/j.tips.2007.06.006.
- 4.
Peterson, S.M.; Pack, T.F.; Wilkins, A.D.; et al. Elucidation of G-Protein and β-Arrestin Functional Selectivity at the Dopamine D2 Receptor. Proc. Natl. Acad. Sci. USA 2015, 112, 7097–7102. https://doi.org/10.1073/pnas.1502742112.
- 5.
Wang, W.; Qiao, Y.; Li, Z.; et al. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 2018, 39, 367–386. https://doi.org/10.1016/j.tips.2018.01.001.
- 6.
Costa-Neto, C.M.; Parreiras-e-Silva, L.T. Deciphering Complexity of GPCR Signaling and Modulation: Implications and Perspectives for Drug Discovery. Clin. Sci. 2025, 139, 463–477. https://doi.org/10.1042/CS20245182.
- 7.
Cvicek, V.; Goddard, W.A. III; Abrol, R. Structure-Based Sequence Alignment of the Transmembrane Domains of All Human GPCRs: Phylogenetic, Structural and Functional Implications. PLoS Comput. Biol. 2016, 12, e1004805. https://doi.org/10.1371/journal.pcbi.1004805.
- 8.
Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. https://doi.org/10.3389/fphar.2019.00125.
- 9.
Lobingier, B.T.; von Zastrow, M. When Trafficking and Signaling Mix: How Subcellular Location Shapes G Protein-Coupled Receptor Activation of Heterotrimeric G Proteins. Traffic 2019, 20, 130–136. https://doi.org/10.1111/tra.12634.
- 10.
Wess, J.; Oteng, A.-B.; Rivera-Gonzalez, O.; et al. β-Arrestins: Structure, Function, Physiology, and Pharmacological Perspectives. Pharmacol. Rev. 2023, 75, 854–884. https://doi.org/10.1124/pharmrev.121.000302.
- 11.
Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J.; et al. Teaching Old Receptors New Tricks: Biasing Seven-Transmembrane Receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. https://doi.org/10.1038/nrd3024.
- 12.
Holobotovskyy, V.; Manzur, M.; Tare, M.; et al. Regulator of G-Protein Signaling 5 Controls Blood Pressure Homeostasis and Vessel Wall Remodeling. Circ. Res. 2013, 112, 781–791. https://doi.org/10.1161/CIRCRESAHA.111.300142.
- 13.
Lymperopoulos, A.; Wertz, S.L.; Pollard, C.M.; et al. Not All Arrestins Are Created Equal: Therapeutic Implications of the Functional Diversity of the β-Arrestins in the Heart. World J. Cardiol. 2019, 11, 47–56. https://doi.org/10.4330/wjc.v11.i2.47.
- 14.
Kim, K.-S.; Abraham, D.; Williams, B.; et al. β-Arrestin-Biased AT1R Stimulation Promotes Cell Survival during Acute Cardiac Injury. Am. J. Physiol.-Heart Circ. Physiol. 2012, 303, H1001–H1010. https://doi.org/10.1152/ajpheart.00475.2012.
- 15.
Teixeira, L.B.; Parreiras-E-Silva, L.T.; Bruder-Nascimento, T.; et al. Ang-(1-7) Is an Endogenous β-Arrestin-Biased Agonist of the AT1 Receptor with Protective Action in Cardiac Hypertrophy. Sci. Rep. 2017, 7, 11903. https://doi.org/10.1038/s41598-017-12074-3.
- 16.
Galandrin, S.; Denis, C.; Boularan, C.; et al. Cardioprotective Angiotensin-(1-7) Peptide Acts as a Natural-Biased Ligand at the Angiotensin II Type 1 Receptor. Hypertension 2016, 68, 1365–1374. https://doi.org/10.1161/HYPERTENSIONAHA.116.08118.
- 17.
Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J.; et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. https://doi.org/10.1016/j.cell.2016.07.004.
- 18.
Nguyen, A.H.; Thomsen, A.R.B.; Cahill, T.J.; et al. Structure of an Endosomal Signaling GPCR-G Protein-β-Arrestin Megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131. https://doi.org/10.1038/s41594-019-0330-y.
- 19.
van Gastel, J.; Hendrickx, J.O.; Leysen, H.; et al. β-Arrestin Based Receptor Signaling Paradigms: Potential Therapeutic Targets for Complex Age-Related Disorders. Front. Pharmacol. 2018, 9, 1369. https://doi.org/10.3389/fphar.2018.01369.
- 20.
Wess, J. The Two β-Arrestins Regulate Distinct Metabolic Processes: Studies with Novel Mutant Mouse Models. Int. J. Mol. Sci. 2022, 23, 495. https://doi.org/10.3390/ijms23010495.
- 21.
Slosky, L.M.; Caron, M.G.; Barak, L.S.; et al. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol. Sci. 2021, 42, 283–299. https://doi.org/10.1016/j.tips.2020.12.005.
- 22.
Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased Signalling: From Simple Switches to Allosteric Microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. https://doi.org/10.1038/nrd.2017.229.
- 23.
Lefkowitz, R.J. A Brief History of G-Protein Coupled Receptors (Nobel Lecture). Angew. Chem. Int. Ed. 2013, 52, 6366–6378. https://doi.org/10.1002/anie.201301924.
- 24.
Milligan, G.; Kostenis, E. Heterotrimeric G-Proteins: A Short History. Br. J. Pharmacol. 2006, 147, S46–S55. https://doi.org/10.1038/sj.bjp.0706405.
- 25.
Avet, C.; Mancini, A.; Breton, B.; et al. Effector Membrane Translocation Biosensors Reveal G Protein and Βarrestin Coupling Profiles of 100 Therapeutically Relevant GPCRs. eLife 2022, 11, e74101. https://doi.org/10.7554/eLife.74101.
- 26.
Hauser, A.S.; Avet, C.; Normand, C.; et al. Common Coupling Map Advances GPCR-G Protein Selectivity. eLife 2022, 11, e74107. https://doi.org/10.7554/eLife.74107.
- 27.
Jelinek, V.; Mösslein, N.; Bünemann, M.; et al. Structures in G Proteins Important for Subtype Selective Receptor Binding and Subsequent Activation. Commun. Biol. 2021, 4, 635. https://doi.org/10.1038/s42003-021-02143-9.
- 28.
Pandey, S.; Saha, S.; Shukla, A.K. Transmitting the Signal: Structure of the β1-Adrenergic Receptor-Gs Protein Complex. Mol. Cell 2020, 80, 3–5. https://doi.org/10.1016/j.molcel.2020.09.016.
- 29.
Cannavo, A.; Rengo, G.; Liccardo, D.; et al. Β1-Adrenergic Receptor and Sphingosine-1-Phosphate Receptor 1 (S1PR1) Reciprocal Downregulation Influences Cardiac Hypertrophic Response and Progression to Heart Failure: Protective Role of S1PR1 Cardiac Gene Therapy. Circulation 2013, 128, 1612–1622. https://doi.org/10.1161/CIRCULATIONAHA.113.002659.
- 30.
Chen, H.; Zhang, S.; Hou, R.; et al. Gi-Protein-Coupled β1-Adrenergic Receptor: Re-Understanding the Selectivity of β1-Adrenergic Receptor to G Protein. Acta Biochim. Biophys. Sin. 2022, 54, 1043–1048. https://doi.org/10.3724/abbs.2022096.
- 31.
Wang, J.; Hanada, K.; Staus, D.P.; et al. Gαi Is Required for Carvedilol-Induced Β1 Adrenergic Receptor β-Arrestin Biased Signaling. Nat. Commun. 2017, 8, 1706. https://doi.org/10.1038/s41467-017-01855-z.
- 32.
Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J.; et al. Switching of the Coupling of the Beta2-Adrenergic Receptor to Different G Proteins by Protein Kinase A. Nature 1997, 390, 88–91. https://doi.org/10.1038/36362.
- 33.
Chesley, A.; Lundberg, M.S.; Asai, T.; et al. The Beta(2)-Adrenergic Receptor Delivers an Antiapoptotic Signal to Cardiac Myocytes through G(i)-Dependent Coupling to Phosphatidylinositol 3’-Kinase. Circ. Res. 2000, 87, 1172–1179. https://doi.org/10.1161/01.res.87.12.1172.
- 34.
Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; et al. Beta-Arrestin-Dependent, G Protein-Independent ERK1/2 Activation by the Beta2 Adrenergic Receptor. J. Biol. Chem. 2006, 281, 1261–1273. https://doi.org/10.1074/jbc.M506576200.
- 35.
Casiraghi, M.; Wang, H.; Brennan, P.; et al. Structure and Dynamics Determine G Protein Coupling Specificity at a Class A GPCR. Sci. Adv. 2025, 11, eadq3971. https://doi.org/10.1126/sciadv.adq3971.
- 36.
Roy, S.; Sinha, S.; Silas, A.J.; et al. Growth Factor-Dependent Phosphorylation of Gαi Shapes Canonical Signaling by G Protein-Coupled Receptors. Sci. Signal. 2024, 17, eade8041. https://doi.org/10.1126/scisignal.ade8041.
- 37.
Dwivedi, H.; Baidya, M.; Shukla, A.K.; et al. GPCR Signaling: The Interplay of Gαi and β-Arrestin. Curr. Biol. 2018, 28, R324–R327. https://doi.org/10.1016/j.cub.2018.02.027.
- 38.
Strohman, M.J.; Maeda, S.; Hilger, D.; et al. Local Membrane Charge Regulates β2 Adrenergic Receptor Coupling to Gi3. Nat. Commun. 2019, 10, 2234. https://doi.org/10.1038/s41467-019-10108-0.
- 39.
Smith, J.S.; Hilibrand, A.S.; Skiba, M.A.; et al. The M3 Muscarinic Acetylcholine Receptor Can Signal through Multiple G Protein Families. Mol. Pharmacol. 2024, 105, 386–394. https://doi.org/10.1124/molpharm.123.000818.
- 40.
Rockman, H.A.; Lefkowitz, R.J. G Protein–Coupled Receptors: From Radioligand Binding to Cellular Signaling. J. Clin. Investig. 2024, 134, e178109. https://doi.org/10.1172/JCI178109.
- 41.
Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; et al. Conformational Biosensors Reveal GPCR Signalling from Endosomes. Nature 2013, 495, 534–538. https://doi.org/10.1038/nature12000.
- 42.
Bénard, G.; Massa, F.; Puente, N.; et al. Mitochondrial CB₁ Receptors Regulate Neuronal Energy Metabolism. Nat. Neurosci. 2012, 15, 558–564. https://doi.org/10.1038/nn.3053.
- 43.
Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; et al. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. https://doi.org/10.1126/science.1106943.
- 44.
Irannejad, R.; Pessino, V.; Mika, D.; et al. Functional Selectivity of GPCR-Directed Drug Action through Location Bias. Nat. Chem. Biol. 2017, 13, 799–806. https://doi.org/10.1038/nchembio.2389.
- 45.
Crilly, S.E.; Puthenveedu, M.A. Compartmentalized GPCR Signaling from Intracellular Membranes. J. Membr. Biol. 2021, 254, 259–271. https://doi.org/10.1007/s00232-020-00158-7.
- 46.
Fasciani, I.; Carli, M.; Petragnano, F.; et al. GPCRs in Intracellular Compartments: New Targets for Drug Discovery. Biomolecules 2022, 12, 1343. https://doi.org/10.3390/biom12101343.
- 47.
Conflitti, P.; Lyman, E.; Sansom, M.S.P.; et al. Functional Dynamics of G Protein-Coupled Receptors Reveal New Routes for Drug Discovery. Nat. Rev. Drug Discov. 2025, 24, 251–275. https://doi.org/10.1038/s41573-024-01083-3.
- 48.
Willette, B.K.A.; Zhang, J.-F.; Zhang, J.; et al. Endosome Positioning Coordinates Spatially Selective GPCR Signaling. Nat. Chem. Biol. 2024, 20, 151–161. https://doi.org/10.1038/s41589-023-01390-7.
- 49.
Agarwal, S.R.; Yang, P.-C.; Rice, M.; et al. Role of Membrane Microdomains in Compartmentation of cAMP Signaling. PLoS ONE 2014, 9, e95835. https://doi.org/10.1371/journal.pone.0095835.
- 50.
Haushalter, K.J.; Casteel, D.E.; Raffeiner, A.; et al. Phosphorylation of Protein Kinase A (PKA) Regulatory Subunit RIα by Protein Kinase G (PKG) Primes PKA for Catalytic Activity in Cells. J. Biol. Chem. 2018, 293, 4411–4421. https://doi.org/10.1074/jbc.M117.809988.
- 51.
Bock, A.; Annibale, P.; Konrad, C.; et al. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 2020, 182, 1519–1530.e17. https://doi.org/10.1016/j.cell.2020.07.035.
- 52.
Agarwal, S.R.; Clancy, C.E.; Harvey, R.D. Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep. 2016, 6, 19577. https://doi.org/10.1038/srep19577.
- 53.
Richards, M.; Lomas, O.; Jalink, K.; et al. Intracellular Tortuosity Underlies Slow cAMP Diffusion in Adult Ventricular Myocytes. Cardiovasc. Res. 2016, 110, 395-407. https://doi.org/10.1093/cvr/cvw080.
- 54.
Shen, A.; Nieves-Cintron, M.; Deng, Y.; et al. Functionally Distinct and Selectively Phosphorylated GPCR Subpopulations Co-Exist in a Single Cell. Nat. Commun. 2018, 9, 1050. https://doi.org/10.1038/s41467-018-03459-7.
- 55.
Shen, A.; Chen, D.; Kaur, M.; et al. β-Blockers Augment L-Type Ca2+ Channel Activity by Targeting Spatially Restricted β2AR Signaling in Neurons. eLife 2019, 8, e49464. https://doi.org/10.7554/eLife.49464.
- 56.
Flores-Espinoza, E.; Thomsen, A.R. Beneath the Surface: Endosomal GPCR Signaling. Trends Biochem. Sci. 2024, 49, 520–531. https://doi.org/10.1016/j.tibs.2024.03.006.
- 57.
Ferrandon, S.; Feinstein, T.N.; Castro, M.; et al. Sustained Cyclic AMP Production by Parathyroid Hormone Receptor Endocytosis. Nat. Chem. Biol. 2009, 5, 734–742. https://doi.org/10.1038/nchembio.206.
- 58.
White, A.D.; Peña, K.A.; Clark, L.J.; et al. Spatial Bias in cAMP Generation Determines Biological Responses to PTH Type 1 Receptor Activation. Sci. Signal. 2021, 14, eabc5944. https://doi.org/10.1126/scisignal.abc5944.
- 59.
Peña, K.A.; White, A.D.; Savransky, S.; et al. Biased GPCR Signaling by the Native Parathyroid Hormone–Related Protein 1 to 141 Relative to Its N-Terminal Fragment 1 to 36. J. Biol. Chem. 2022, 298, 102332. https://doi.org/10.1016/j.jbc.2022.102332.
- 60.
Tsvetanova, N.G.; von Zastrow, M. Spatial Encoding of Cyclic AMP Signaling Specificity by GPCR Endocytosis. Nat. Chem. Biol. 2014, 10, 1061–1065. https://doi.org/10.1038/nchembio.1665.
- 61.
Teixeira, L.B.; Blouin, M.-J.; Le Gouill, C.; et al. Sustained Gαs Signaling Mediated by Vasopressin Type 2 Receptors Is Ligand Dependent but Endocytosis and β-Arrestin Independent. Sci. Signal. 2025, 18, eadf6206. https://doi.org/10.1126/scisignal.adf6206.
- 62.
Blythe, E.E.; von Zastrow, M. β-Arrestin-Independent Endosomal cAMP Signaling by a Polypeptide Hormone GPCR. Nat. Chem. Biol. 2024, 20, 323–332. https://doi.org/10.1038/s41589-023-01412-4.
- 63.
Xu, B.; Wang, Y.; Bahriz, S.M.; et al. Probing Spatiotemporal PKA Activity at the Ryanodine Receptor and SERCA2a Nanodomains in Cardomyocytes. Cell Commun. Signal. 2022, 20, 143. https://doi.org/10.1186/s12964-022-00947-8.
- 64.
Wang, Y.; Shi, Q.; Li, M.; et al. Intracellular Β1-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility. Circ. Res. 2021, 128, 246–261. https://doi.org/10.1161/CIRCRESAHA.120.317452.
- 65.
Wang, Y.; Zhao, M.; Xu, B.; et al. Monoamine Oxidase A and Organic Cation Transporter 3 Coordinate Intracellular β1AR Signaling to Calibrate Cardiac Contractile Function. Basic Res. Cardiol. 2022, 117, 37. https://doi.org/10.1007/s00395-022-00944-5.
- 66.
Wang, Y.; Zhao, M.; Shi, Q.; et al. Monoamine Oxidases Desensitize Intracellular β1AR Signaling in Heart Failure. Circ. Res. 2021, 129, 965–967. https://doi.org/10.1161/CIRCRESAHA.121.319546.
- 67.
Fu, Q.; Wang, Y.; Yan, C.; et al. Phosphodiesterase in Heart and Vessels: From Physiology to Diseases. Physiol. Rev. 2024, 104, 765–834. https://doi.org/10.1152/physrev.00015.2023.
- 68.
Benton, K.C.; Wheeler, D.S.; Kurtoglu, B.; et al. Norepinephrine Activates β1-adrenergic Receptors at the Inner Nuclear Membrane in Astrocytes. Glia 2022, 70, 1777–1794. https://doi.org/10.1002/glia.24219.
- 69.
Kunselman, J.M.; Lott, J.; Puthenveedu, M.A. Mechanisms of Selective G Protein–Coupled Receptor Localization and Trafficking. Curr. Opin. Cell Biol. 2021, 71, 158–165. https://doi.org/10.1016/j.ceb.2021.03.002.
- 70.
Lin, T.-Y.; Mai, Q.N.; Zhang, H.; et al. Cardiac Contraction and Relaxation Are Regulated by Distinct Subcellular cAMP Pools. Nat. Chem. Biol. 2024, 20, 62–73. https://doi.org/10.1038/s41589-023-01381-8.
- 71.
Duarte, M.L.; Devi, L.A. Post-Translational Modifications of Opioid Receptors. Trends Neurosci. 2020, 43, 417–432. https://doi.org/10.1016/j.tins.2020.03.011.
- 72.
Tang, X.; Bian, J.; Li, Z. Posttranslational Modifications in GPCR Internalization. Am. J. Physiol. Cell Physiol. 2022, 323, C84–C94. https://doi.org/10.1152/ajpcell.00015.2022.
- 73.
Cahill, T.J.; Thomsen, A.R.B.; Tarrasch, J.T.; et al. Distinct Conformations of GPCR-β-Arrestin Complexes Mediate Desensitization, Signaling, and Endocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567. https://doi.org/10.1073/pnas.1701529114.
- 74.
Wu, J.-J.; Yang, Y.; Peng, W.-T.; et al. G Protein-Coupled Receptor Kinase 2 Regulating β2-Adrenergic Receptor Signaling in M2-Polarized Macrophages Contributes to Hepatocellular Carcinoma Progression. OncoTargets Ther. 2019, 12, 5499–5513. https://doi.org/10.2147/OTT.S209291.
- 75.
Tran, T.; Friedman, J.; Qunaibi, E.; et al. Characterization of Agonist Stimulation of cAMP-Dependent Protein Kinase and G Protein-Coupled Receptor Kinase Phosphorylation of the Beta2-Adrenergic Receptor Using Phosphoserine-Specific Antibodies. Mol. Pharmacol. 2004, 65, 196–206. https://doi.org/10.1124/mol.65.1.196.
- 76.
Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; et al. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 2020, 183, 1813–1825.e18. https://doi.org/10.1016/j.cell.2020.11.014.
- 77.
Zhang, F.; Steinberg, S.F. S49G and R389G Polymorphisms of the β1-Adrenergic Receptor Influence Signaling via the cAMP-PKA and ERK Pathways. Physiol. Genom. 2013, 45, 1186–1192. https://doi.org/10.1152/physiolgenomics.00087.2013.
- 78.
Ahles, A.; Rodewald, F.; Rochais, F.; et al. Interhelical Interaction and Receptor Phosphorylation Regulate the Activation Kinetics of Different Human β1-Adrenoceptor Variants. J. Biol. Chem. 2015, 290, 1760–1769. https://doi.org/10.1074/jbc.M114.607333.
- 79.
Levin, M.C.; Marullo, S.; Muntaner, O.; et al. The Myocardium-Protective Gly-49 Variant of the β1-Adrenergic Receptor Exhibits Constitutive Activity and Increased Desensitization and Down-Regulation. J. Biol. Chem. 2002, 277, 30429–30435. https://doi.org/10.1074/jbc.M200681200.
- 80.
Schjoldager, K.T.-B.G.; Clausen, H. Site-Specific Protein O-Glycosylation Modulates Proprotein Processing—Deciphering Specific Functions of the Large Polypeptide GalNAc-Transferase Gene Family. Biochim. Biophys. Acta 2012, 1820, 2079–2094. https://doi.org/10.1016/j.bbagen.2012.09.014.
- 81.
Tuhkanen, H.E.; Haasiomäki, I.J.; Lackman, J.J.; et al. Altered O‐glycosylation of β1‐adrenergic Receptor N‐terminal Single‐nucleotide Variants Modulates Receptor Processing and Functional Activity. FEBS J. 2025, 292, 998–1018. https://doi.org/10.1111/febs.17257.
- 82.
Palczewski, K. Oligomeric Forms of G Protein-Coupled Receptors (GPCRs). Trends Biochem. Sci. 2010, 35, 595–600. https://doi.org/10.1016/j.tibs.2010.05.002.
- 83.
Farran, B.; et al. An Update on the Physiological and Therapeutic Relevance of GPCR Oligomers. Pharmacol. Res. 2017, 117, 303–327. https://doi.org/10.1016/j.phrs.2017.01.008.
- 84.
Liu, J.; Tang, H.; Xu, C.; et al. Biased Signaling Due to Oligomerization of the G Protein-Coupled Platelet-Activating Factor Receptor. Nat. Commun. 2022, 13, 6365. https://doi.org/10.1038/s41467-022-34056-4.
- 85.
Delille, H.K.; Becker, J.M.; Burkhardt, S.; et al. Heterocomplex Formation of 5-HT2A-mGlu2 and Its Relevance for Cellular Signaling Cascades. Neuropharmacology 2012, 62, 2184–2191. https://doi.org/10.1016/j.neuropharm.2012.01.010.
- 86.
Harishkumar, R.; Hans, S.; Stanton, J.E.; et al. Targeting the Platelet-Activating Factor Receptor (PAF-R): Antithrombotic and Anti-Atherosclerotic Nutrients. Nutrients 2022, 14, 4414. https://doi.org/10.3390/nu14204414.
- 87.
Tsoupras, A.; Adamantidi, T.; Finos, M.A.; et al. Re-Assessing the Role of Platelet Activating Factor and Its Inflammatory Signaling and Inhibitors in Cancer and Anti-Cancer Strategies. Front. Biosci. 2024, 29, 345. https://doi.org/10.31083/j.fbl2910345.
- 88.
Moreno, J.L.; Miranda-Azpiazu, P.; García-Bea, A.; et al. Allosteric Signaling through an mGlu2 and 5-HT2A Heteromeric Receptor Complex and Its Potential Contribution to Schizophrenia. Sci. Signal. 2016, 9, ra5. https://doi.org/10.1126/scisignal.aab0467.
- 89.
Bécamel, C.; Berthoux, C.; Barre, A.; et al. Growing Evidence for Heterogeneous Synaptic Localization of 5-HT2A Receptors. ACS Chem. Neurosci. 2017, 8, 897–899. https://doi.org/10.1021/acschemneuro.6b00409.
- 90.
Song, Y.; Xu, C.; Liu, J.; et al. Heterodimerization With 5-HT2BR Is Indispensable for β2AR-Mediated Cardioprotection. Circ. Res. 2021, 128, 262–277. https://doi.org/10.1161/CIRCRESAHA.120.317011.
- 91.
Bai, B.; Liu, L.; Zhang, N.; et al. Heterodimerization of Human Apelin and Bradykinin 1 Receptors: Novel Signal Transduction Characteristics. Cell. Signal. 2014, 26, 1549–1559. https://doi.org/10.1016/j.cellsig.2014.03.022.
- 92.
Rozenfeld, R.; Devi, L.A. Receptor Heterodimerization Leads to a Switch in Signaling: Beta-Arrestin2-Mediated ERK Activation by Mu-Delta Opioid Receptor Heterodimers. FASEB J. 2007, 21, 2455–2465. https://doi.org/10.1096/fj.06-7793com.
- 93.
Daniel, S.K.; Seo, Y.D.; Pillarisetty, V.G.; et al. The CXCL12-CXCR4/CXCR7 Axis as a Mechanism of Immune Resistance in Gastrointestinal Malignancies. Semin. Cancer Biol. 2020, 65, 176–188. https://doi.org/10.1016/j.semcancer.2019.12.007.
- 94.
Shin, E.; Ko, K.S.; Rhee, B.D.; et al. Different Effects of Prolonged β-Adrenergic Stimulation on Heart and Cerebral Artery. Integr. Med. Res. 2014, 3, 204–210. https://doi.org/10.1016/j.imr.2014.10.002.
- 95.
Lohse, M.J.; Engelhardt, S.; Eschenhagen, T.; et al. What Is the Role of Beta-Adrenergic Signaling in Heart Failure? Circ. Res. 2003, 93, 896–906. https://doi.org/10.1161/01.RES.0000102042.83024.CA.
- 96.
Joca, H.C.; Santos‐Miranda, A.; Joviano-Santos, J.V.; et al. Chronic Sympathetic Hyperactivity Triggers Electrophysiological Remodeling and Disrupts Excitation-Contraction Coupling in Heart. Sci. Rep. 2020, 10, 8001. https://doi.org/10.1038/s41598-020-64949-7.
- 97.
Düngen, H.-D.; Dordevic, A.; Felix, S.B.; et al. β1-Adrenoreceptor Autoantibodies in Heart Failure: Physiology and Therapeutic Implications. Circ. Heart Fail. 2020, 13, e006155. https://doi.org/10.1161/CIRCHEARTFAILURE.119.006155.
- 98.
Madamanchi, A. β-Adrenergic Receptor Signaling in Cardiac Function and Heart Failure. McGill J. Med. 2007, 10, 99–104.
- 99.
Manrique, C.; Giles, T.D.; Ferdinand, K.C.; et al. Realities of Newer β‐Blockers for the Management of Hypertension. J. Clin. Hypertens. 2009, 11, 369–375. https://doi.org/10.1111/j.1751-7176.2009.00140.x.
- 100.
Ray, S.; Nair, T.; Sawhney, J.; et al. Role of β-Blockers in the Cardiovascular Disease Continuum: A Collaborative Delphi Survey-Based Consensus from Asia-Pacific. Curr. Med. Res. Opin. 2023, 39, 1671–1683. https://doi.org/10.1080/03007995.2023.2256218.
- 101.
Masarone, D.; Martucci, M.L.; Errigo, V.; et al. The Use of β-Blockers in Heart Failure with Reduced Ejection Fraction. J. Cardiovasc. Dev. Dis. 2021, 8, 101. https://doi.org/10.3390/jcdd8090101.
- 102.
Benkel, T.; Zimmermann, M.; Zeiner, J.; et al. How Carvedilol Activates Β2-Adrenoceptors. Nat. Commun. 2022, 13, 7109. https://doi.org/10.1038/s41467-022-34765-w.
- 103.
Wang, Q.; Wang, Y.; West, T.M.; et al. Carvedilol Induces Biased Β1 Adrenergic Receptor-Nitric Oxide Synthase 3-Cyclic Guanylyl Monophosphate Signalling to Promote Cardiac Contractility. Cardiovasc. Res. 2021, 117, 2237–2251. https://doi.org/10.1093/cvr/cvaa266.
- 104.
Walweel, K.; Cheesman, E.; Molenaar, P.; et al. Potential of β2AR for Added Benefit in Treating Heart Failure through a Better Understanding of Signaling. Curr. Opin. Physiol. 2023, 36, 100719. https://doi.org/10.1016/j.cophys.2023.100719.
- 105.
Fajardo, G.; Zhao, M.; Berry, G.; et al. β2-Adrenergic Receptors Mediate Cardioprotection through Crosstalk with Mitochondrial Cell Death Pathways. J. Mol. Cell. Cardiol. 2011, 51, 781–789. https://doi.org/10.1016/j.yjmcc.2011.06.019.
- 106.
Carr, R.; Schilling, J.; Song, J.; et al. β-Arrestin-Biased Signaling through the β2-Adrenergic Receptor Promotes Cardiomyocyte Contraction. Proc. Natl. Acad. Sci. USA 2016, 113, E4107–E4116. https://doi.org/10.1073/pnas.1606267113.
- 107.
Lamichhane, R.; Liu, J.J.; White, K.L.; et al. Biased Signaling of the G-Protein-Coupled Receptor β2AR Is Governed by Conformational Exchange Kinetics. Structure 2020, 28, 371–377.e3. https://doi.org/10.1016/j.str.2020.01.001.
- 108.
Maslov, L.N.; Naryzhnaya, N.V.; Voronkov, N.S.; et al. The Role of β-Adrenergic Receptors in the Regulation of Cardiac Tolerance to Ischemia/Reperfusion. Why Do β-Adrenergic Receptor Agonists and Antagonists Protect the Heart? Fundam. Clin. Pharmacol. 2024, 38, 658–673. https://doi.org/10.1111/fcp.12988.
- 109.
Odnoshivkina, Y.G.; Petrov, A.M.; Zefirov, A.L.; et al. The Effects of β2-Adrenoreceptor Activation on the Contractility, Ca-Signals and Nitric Oxide Production in the Mouse Atria. Acta Nat. 2011, 3, 103–112.
- 110.
Ahmet, I.; Turner, T.; Lakatta, E.G.; et al. Fenoterol Enantiomers Do Not Possess Beneficial Therapeutic Properties of Their Racemic Mixture in the Rat Model of Post Myocardial Infarction Dilated Cardiomyopathy. Cardiovasc. Drugs Ther. 2012, 26, 101–108. https://doi.org/10.1007/s10557-011-6366-9.
- 111.
Castoldi, G.; Carletti, R.; Ippolito, S.; et al. Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension. Int. J. Mol. Sci. 2021, 22, 13678. https://doi.org/10.3390/ijms222413678.
- 112.
Zhong, J.; Basu, R.; Guo, D.; et al. Angiotensin-Converting Enzyme 2 Suppresses Pathological Hypertrophy, Myocardial Fibrosis, and Cardiac Dysfunction. Circulation 2010, 122, 717–728. https://doi.org/10.1161/CIRCULATIONAHA.110.955369.
- 113.
Singh, K.D.; Karnik, S.S.; et al. Implications of β-Arrestin Biased Signaling by Angiotensin II Type 1 Receptor for Cardiovascular Drug Discovery and Therapeutics. Cell. Signal. 2024, 124, 111410. https://doi.org/10.1016/j.cellsig.2024.111410.
- 114.
Cao, Y.; Kumar, S.; Namkung, Y.; et al. Angiotensin II Type 1 Receptor Variants Alter Endosomal Receptor-β-Arrestin Complex Stability and MAPK Activation. J. Biol. Chem. 2020, 295, 13169–13180. https://doi.org/10.1074/jbc.RA120.014330.
- 115.
Cao, Y.; van der Velden, W.J.C.; Namkung, Y.; et al. Unraveling Allostery within the Angiotensin II Type 1 Receptor for Gαq and β-Arrestin Coupling. Sci. Signal. 2023, 16, eadf2173. https://doi.org/10.1126/scisignal.adf2173.
- 116.
Violin, J.D.; DeWire, S.M.; Yamashita, D.; et al. Selectively Engaging β-Arrestins at the Angiotensin II Type 1 Receptor Reduces Blood Pressure and Increases Cardiac Performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. https://doi.org/10.1124/jpet.110.173005.
- 117.
Monasky, M.M.; Taglieri, D.M.; Henze, M.; et al. The β-Arrestin-Biased Ligand TRV120023 Inhibits Angiotensin II-Induced Cardiac Hypertrophy While Preserving Enhanced Myofilament Response to Calcium. Am. J. Physiol. -Heart Circ. Physiol. 2013, 305, H856. https://doi.org/10.1152/ajpheart.00327.2013.
- 118.
Boerrigter, G.; Soergel, D.G.; Violin, J.D.; et al. TRV120027, a Novel β-Arrestin Biased Ligand at the Angiotensin II Type I Receptor, Unloads the Heart and Maintains Renal Function When Added to Furosemide in Experimental Heart Failure. Circ. Heart Fail. 2012, 5, 627–634. https://doi.org/10.1161/CIRCHEARTFAILURE.112.969220.
- 119.
Dinh, W.; Albrecht-Küpper, B.; Gheorghiade, M.; et al. Partial Adenosine A1 Agonist in Heart Failure. In Heart Failure; Springer: Cham, Switzerland, 2016; Volume 243, pp. 177–203. https://doi.org/10.1007/164_2016_83.
- 120.
Valant, C.; May, L.T.; Aurelio, L.; et al. Separation of On-Target Efficacy from Adverse Effects through Rational Design of a Bitopic Adenosine Receptor Agonist. Proc. Natl. Acad. Sci. USA 2014, 111, 4614–4619. https://doi.org/10.1073/pnas.1320962111.
- 121.
Rueda, P.; Merlin, J.; Chimenti, S.; et al. Pharmacological Insights into Safety and Efficacy Determinants for the Development of Adenosine Receptor Biased Agonists in the Treatment of Heart Failure. Front. Pharmacol. 2021, 12, 628060. https://doi.org/10.3389/fphar.2021.628060.
- 122.
Shah, S.J.; Voors, A.A.; McMurray, J.J.V.; et al. Effect of Neladenoson Bialanate on Exercise Capacity Among Patients with Heart Failure with Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA 2019, 321, 2101–2112. https://doi.org/10.1001/jama.2019.6717.
- 123.
Voors, A.A.; Bax, J.J.; Hernandez, A.F.; et al. Safety and Efficacy of the Partial Adenosine A1 Receptor Agonist Neladenoson Bialanate in Patients with Chronic Heart Failure with Reduced Ejection Fraction: A Phase IIb, Randomized, Double-Blind, Placebo-Controlled Trial. Eur. J. Heart Fail. 2019, 21, 1426–1433. https://doi.org/10.1002/ejhf.1591.
- 124.
Chapman, F.A.; Maguire, J.J.; Newby, D.E.; et al. Targeting the Apelin System for the Treatment of Cardiovascular Diseases. Cardiovasc. Res. 2023, 119, 2683–2696. https://doi.org/10.1093/cvr/cvad171.
- 125.
Brame, A.L.; Maguire, J.J.; Yang, P.; et al. Design, Characterization, and First-in-Human Study of the Vascular Actions of a Novel Biased Apelin Receptor Agonist. Hypertension 2015, 65, 834–840. https://doi.org/10.1161/HYPERTENSIONAHA.114.05099.
- 126.
Coquerel, D.; Delile, E.; Dumont, L.; et al. Gαi-Biased Apelin Analog Protects against Isoproterenol-Induced Myocardial Dysfunction in Rats. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H1646–H1656. https://doi.org/10.1152/ajpheart.00688.2020.
- 127.
Wang, W.-W.; Ji, S.-Y.; Zhang, W.; et al. Structure-Based Design of Non-Hypertrophic Apelin Receptor Modulator. Cell 2024, 187, 1460–1475.e20. https://doi.org/10.1016/j.cell.2024.02.004.
- 128.
Lamberts, J.T.; Traynor, J.R. Opioid Receptor Interacting Proteins and the Control of Opioid Signaling. Curr. Pharm. Des. 2013, 19, 7333–7347. https://doi.org/10.2174/138161281942140105160625.
- 129.
Friedman, A.; Nabong, L. Opioids: Pharmacology, Physiology, and Clinical Implications in Pain Medicine. Phys. Med. Rehabil. Clin. N. Am. 2020, 31, 289–303. https://doi.org/10.1016/j.pmr.2020.01.007.
- 130.
Paul, A.K.; Smith, C.M.; Rahmatullah, M.; et al. Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, 1091. https://doi.org/10.3390/ph14111091.
- 131.
Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; et al. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e13. https://doi.org/10.1016/j.cell.2017.10.035.
- 132.
Ma, M.; Sun, J.; Li, M.; et al. Synthesis and Evaluation of Novel Biased μ-Opioid-Receptor (μOR) Agonists. Molecules 2019, 24, 259. https://doi.org/10.3390/molecules24020259.
- 133.
Gillis, A.; Gondin, A.B.; Kliewer, A.; et al. Low Intrinsic Efficacy for G Protein Activation Can Explain the Improved Side Effect Profiles of New Opioid Agonists. Sci. Signal. 2020, 13, eaaz3140. https://doi.org/10.1126/scisignal.aaz3140.
- 134.
Ramos-Gonzalez, N.; Paul, B.; Majumdar, S.; et al. IUPHAR Themed Review: Opioid Efficacy, Bias, and Selectivity. Pharmacol. Res. 2023, 197, 106961. https://doi.org/10.1016/j.phrs.2023.106961.
- 135.
Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. https://doi.org/10.3389/fphar.2020.01003.
- 136.
Allen, J.A.; Yost, J.M.; Setola, V.; et al. Discovery of β-Arrestin-Biased Dopamine D2 Ligands for Probing Signal Transduction Pathways Essential for Antipsychotic Efficacy. Proc. Natl. Acad. Sci. USA 2011, 108, 18488–18493. https://doi.org/10.1073/pnas.1104807108.
- 137.
Yano, H.; Cai, N.-S.; Xu, M.; et al. Gs- versus Golf-Dependent Functional Selectivity Mediated by the Dopamine D1 Receptor. Nat. Commun. 2018, 9, 486. https://doi.org/10.1038/s41467-017-02606-w.
- 138.
Duan, W.; Cao, D.; Wang, S.; et al. Serotonin 2A Receptor (5-HT2AR) Agonists: Psychedelics and Non-Hallucinogenic Analogues as Emerging Antidepressants. Chem. Rev. 2024, 124, 124–163. https://doi.org/10.1021/acs.chemrev.3c00375.
- 139.
Cao, D.; Yu, J.; Wang, H.; et al. Structure-Based Discovery of Nonhallucinogenic Psychedelic Analogs. Science 2022, 375, 403–411. https://doi.org/10.1126/science.abl8615.
- 140.
Wallach, J.; Cao, A.B.; Calkins, M.M.; et al. Identification of 5-HT2A Receptor Signaling Pathways Associated with Psychedelic Potential. Nat. Commun. 2023, 14, 8221. https://doi.org/10.1038/s41467-023-44016-1.
- 141.
Fan, L.; Wang, S.; et al. Biased GPCR Signaling: Possible Mechanisms and Therapeutic Applications. Biochemistry 2025, 64, 1180–1192. https://doi.org/10.1021/acs.biochem.4c00827.
- 142.
Wootten, D.; Christopoulos, A.; Marti-Solano, M.; et al. Mechanisms of Signalling and Biased Agonism in G Protein-Coupled Receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. https://doi.org/10.1038/s41580-018-0049-3.
- 143.
Zhang, M.; Chen, T.; Lu, X.; et al. G Protein-Coupled Receptors (GPCRs): Advances in Structures, Mechanisms and Drug Discovery. Signal Transduct. Target. Ther. 2024, 9, 88. https://doi.org/10.1038/s41392-024-01803-6.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com