Cardiovascular disease remains the leading global cause of death, with rising prevalence and mortality, underscoring the need for regenerative strategies. This review synthesizes evidence on mesenchymal stromal/stem cells and their exosomes in cardiac repair. Preclinical studies show paracrine-driven benefits such as pro-angiogenic, anti-apoptotic, anti-fibrotic, and immunomodulatory effects, which may be driven in part by exosomal cargo such as microRNAs. Early clinical trials establish safety and signal improvements in remodeling and function, however, Phase III trial results have been mixed, and no MSC-based product has yet been approved for routine clinical use. We also discuss next-generation induced pluripotent stem cells-derived mesenchymal stromal/stem cells, which offer scalability and consistency but require rigorous control of residual pluripotency and tumorigenicity risk. Future priorities include standardized GMP manufacturing, source and payload optimization, targeted delivery (including cell-free exosomes and biomaterials), and biomarker-guided patient selection to enable effective clinical translation.
- Open Access
- Review
Mesenchymal Stem Cells and Their Exosomes in Cardiac Repair: Advances and Future Directions
- Zhijun Wang 1,
- Na Gao 2,
- Jiandong Zhang 1,
- Baoqiang Guo 1,*
Author Information
Received: 23 Sep 2025 | Revised: 31 Dec 2025 | Accepted: 29 Jan 2026 | Published: 26 Jun 2026
Abstract
Keywords
mesenchymal stem cells | iPSCs | exosomes | cardiac repair | immunomodulation
1. Introduction
Cardiovascular disease (CVD), encompassing ischemic heart disease (IHD) and heart failure (HF), remains the leading cause of mortality worldwide. Data from the Global Burden of Disease (GBD) study indicate that CVD prevalence has risen substantially over the past three decades, and this trend is projected to continue, driven in part by global population aging [1,2,3]. Beyond its health toll, CVD imposes substantial economic costs through medical care and productivity loss [4].
This considerable mortality and disability burden reflects the complex pathophysiology of CVD. In IHD, abrupt interruption of coronary blood flow triggers acute myocardial ischemia and hypoxia; in contrast, HF often represents the chronic end stage of diverse cardiac disorders and is characterized by structural remodeling and impaired contractility [5,6]. In both settings, adult cardiomyocytes have minimal regenerative capacity—lost myocardium is not replaced and cardiac function cannot fully recover after injury. Current standard therapies, including pharmacotherapy, percutaneous coronary intervention (PCI), and coronary artery bypass grafting (CABG) have improved outcomes but cannot restore lost myocardium. Moreover, heart transplantation and left ventricular assist devices (LVADs) are constrained by donor scarcity, immune rejection, and high costs [7,8,9]. The irreversible loss of cardiomyocytes therefore creates an urgent unmet need for therapies capable of regenerating functional heart tissue [10].
Against this backdrop, mesenchymal stromal/stem cell (MSC) therapy has emerged as a promising frontier in cardiac medicine. However, despite compelling paracrine-mediated cardioprotection in preclinical models, clinical translation has remained inconsistent—likely reflecting low myocardial retention, product heterogeneity, and a mismatch between MSCs’ dominant mechanistic actions (notably immunomodulation) and the structural endpoints commonly used in trials (e.g., LVEF). This review provides a comprehensive assessment of the current landscape of MSC-based cardiac repair. We first summarize MSC biology and the mechanisms underlying their reparative effects in the heart. We then critically evaluate evidence from recent clinical trials to assess therapeutic efficacy. Finally, we discuss key translational barriers and emerging strategies to enhance MSC therapy, and conclude with future perspectives on its clinical translation.
2. Biology of MSC
MSCs are adult progenitors with multilineage differentiation potential. First described in the 1970s by Friedenstein and colleagues in bone marrow, they were noted for self-renewal during serial expansion and for differentiating, under defined conditions, into osteoblasts and adipocytes and later also chondrocytes. More recently, MSCs have been induced to exhibit functional cardiomyocyte-like and endothelial-like phenotypes, further extending their reported differentiation repertoire [11,12]. In the 1990s, Caplan popularized the term “mesenchymal stem cell” [13,14]. MSCs can be isolated from a broad range of adult and perinatal tissues. Bone marrow and adipose tissue remain the most frequently used sources in clinical studies, while alternative sources—including dental pulp, umbilical cord and perinatal tissues (e.g., placenta and amniotic-derived compartments), synovium, and lung-resident MSC populations—are increasingly explored to address source-specific heterogeneity (Figure 1) [15,16,17,18,19,20,21]. Beyond their robust multipotency, MSCs exhibit potent paracrine and immunoregulatory activities. Depending on their tissue of origin and microenvironmental cues, MSCs secrete a broad repertoire of bioactive mediators—such as cytokines, chemokines, growth factors, extracellular vesicles, and matrix-remodeling enzymes—which collectively promote angiogenesis, limit apoptosis, and modulate immune responses, thereby enhancing endogenous repair programs and improving organ function. Increasing evidence supports paracrine signaling as a principal driver of MSC-mediated tissue repair [22,23,24,25]. In addition, MSCs can modulate immune responses through both secreted factors and direct interactions with immune cells, underpinning their broad translational promise across disciplines [26].
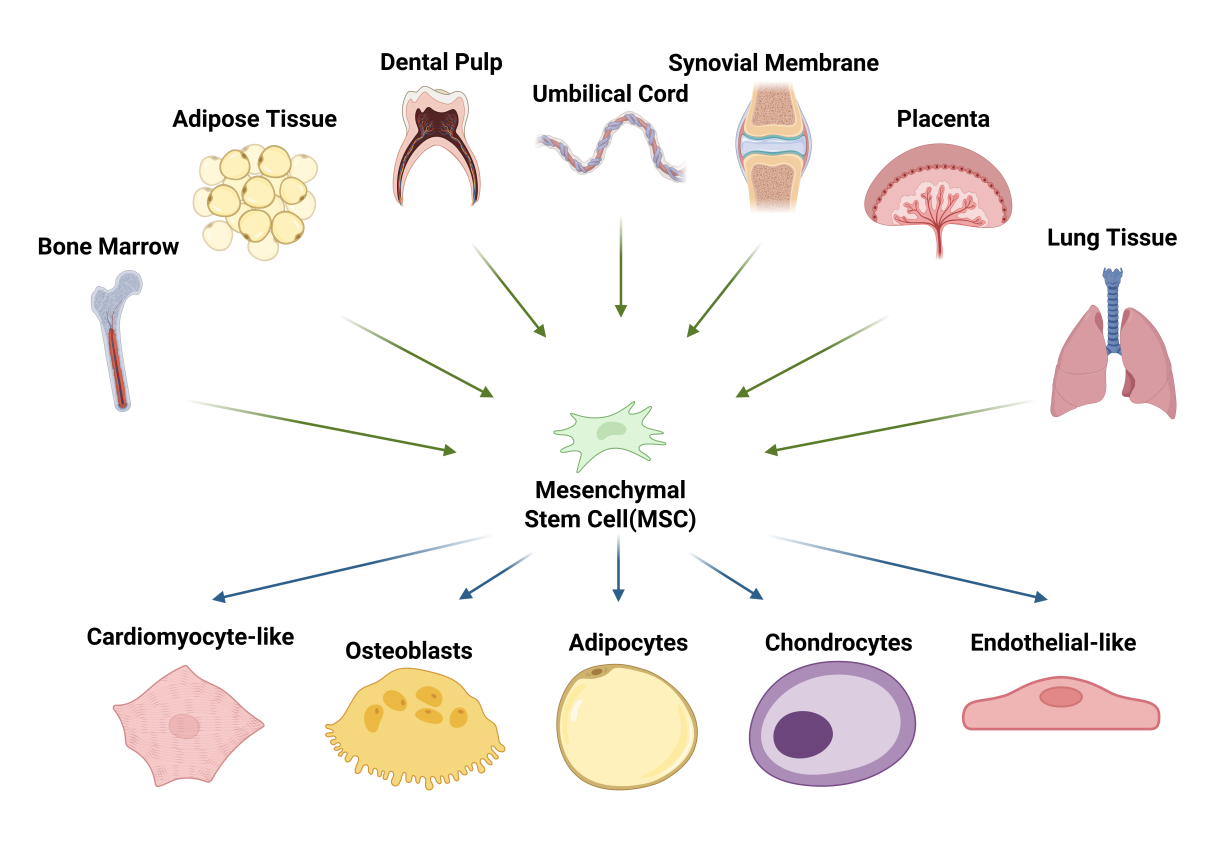
In addition to their in vitro expansion capacity and multilineage differentiation potential, MSCs exhibit a characteristic immunophenotypic profile. According to the minimal criteria proposed by the International Society for Cellular Therapy (ISCT), MSCs should (i) be plastic-adherent under standard culture conditions; (ii) express CD105, CD73, and CD90 while lacking expression of hematopoietic and endothelial markers, including CD45, CD34, CD14 or CD11b, CD79a or CD19, and HLA-DR; and (iii) differentiate in vitro into osteoblasts, chondrocytes, and adipocytes [27]. These harmonized criteria have improved comparability across laboratories and provide a foundation for clinical-grade manufacturing, quality control, and regulatory oversight.
3. Mechanisms of MSCs and MSC-Derived Exosomes in Cardiac Repair
Because of their multilineage differentiation potential, immunomodulatory activity, and relative ease of isolation, MSCs have attracted substantial interest for cardiac repair and are widely studied for the treatment of acute myocardial infarction (MI) and chronic IHD [28,29,30]. In the setting of cardiac injury where cardiomyocyte loss, inflammation, and impaired vascularization coexist, MSCs may contribute to repair through limited lineage commitment, secretion of pro-angiogenic and cytoprotective factors, and immunomodulatory actions that collectively shape a pro-reparative milieu. Recent studies suggest that the therapeutic benefits of MSCs in cardiac repair are mediated predominantly by paracrine signaling rather than direct differentiation into cardiomyocytes, with exosomes serving as key effectors of these mechanisms [31,32,33,34]. These nanoscale extracellular vesicles carry bioactive proteins and nucleic acids, after uptake by recipient cells, they can modulate multiple biological programs, recapitulating key paracrine effects of their parent MSCs and recapitulating key paracrine actions of MSCs and thereby orchestrating multi-cellular repair responses in preclinical models [35,36,37,38,39,40,41]. Compared with whole-cell MSC therapy, exosomes have been reported to show better myocardial retention, a lower risk of immune rejection, and, in some settings, even greater cardioprotective efficacy [42,43,44].
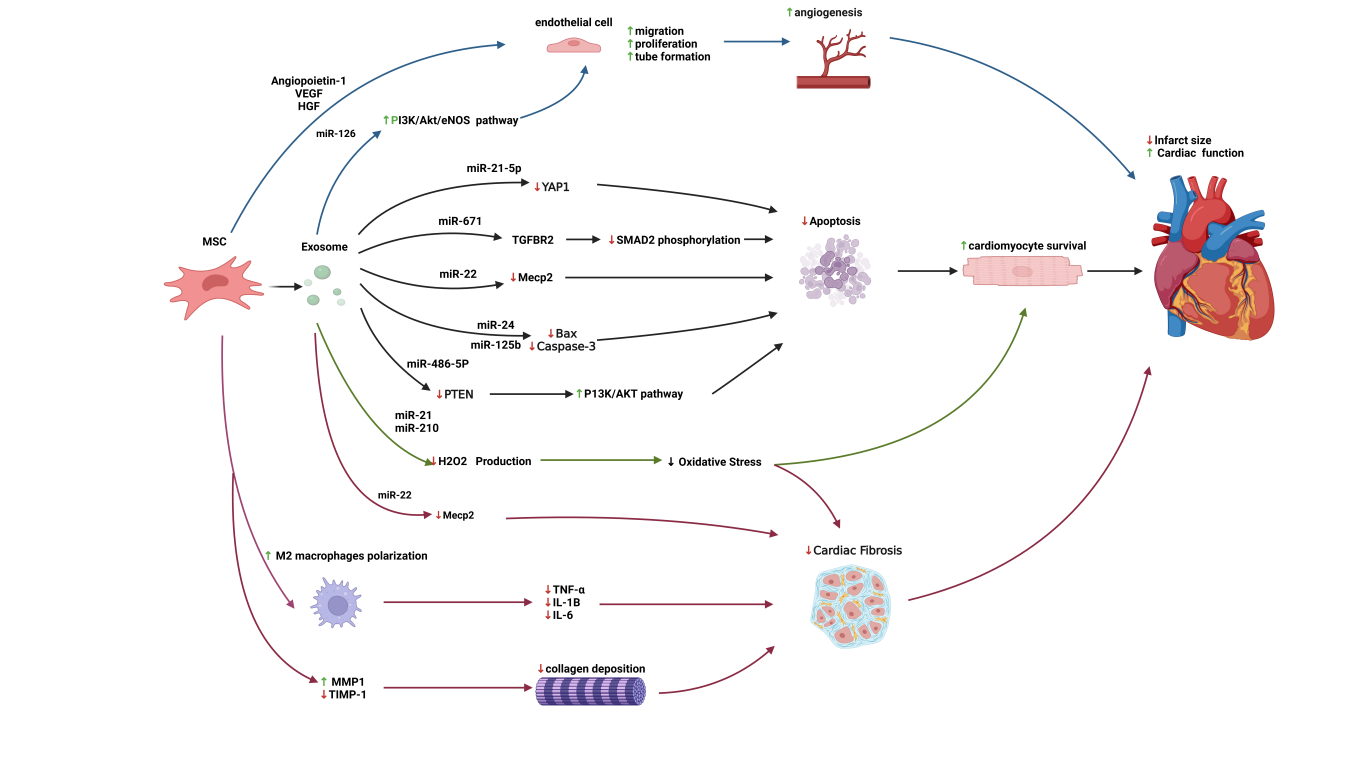
Accordingly, the following sections discuss angiogenesis, cytoprotection, and anti-fibrotic remodeling as core outcomes of MSC paracrine signaling, with MSC-Exos as the principal executors (Figure 2).
3.1. Angiogenesis
MSCs from diverse tissue sources exhibit strong pro-angiogenic properties and promote neovascularization through multiple mechanisms, thereby improving perfusion and the local metabolic milieu within ischemic regions and facilitating the recovery and remodeling of injured myocardium [45]. Transplanted MSCs can home to sites of myocardial injury and, in some studies, have been reported to adopt endothelial- or vascular smooth muscle-like phenotypes; however, they also drive neovascularization through secretion of a broad spectrum of pro-angiogenic and cytoprotective mediators (e.g., VEGF, HGF, and chemokines such as SDF-1α). These factors enhance endothelial survival and sprouting, support vascular maturation, and ultimately improve post-ischemic neovascularization and cardiac function [34,46,47].
In a clinically relevant porcine model of chronic ischemic cardiomyopathy, Tang and colleagues reported that repeated intravenous infusions of allogeneic MSCs improved left ventricular function and structure and reduced myocardial hypertrophy and fibrosis, supporting a sustained pro-reparative effect in vivo [48]. Consistent with a central role for VEGF signaling in MSC-mediated vascular repair, transient mRNA-based augmentation of VEGFA (with bFGF) in human adipose-derived stromal/stem cells enhanced vascular network stability at the transplantation site and improved post-MI cardiac function [49].
MSC-Exos have likewise been shown to carry a repertoire of pro-angiogenic microRNAs; for example, miR-126–enriched vesicles activate the PI3K–Akt–eNOS signaling axis and promote endothelial tube formation under ischemia–reperfusion (I/R) conditions [50]. Recent studies further highlight the role of miR-210 in promoting angiogenesis and metabolic reprogramming in hypoxic environments by upregulating GLUT1 and PDK1 while suppressing fatty acid oxidation enzymes like CPT1 and CD36 [41]. Consistent with these pro-angiogenic actions across ischemic tissues, clinical studies in diabetic foot ulcers (DFU) suggest that MSC-secreted factors, particularly angiopoietin-1 (Ang-1) mobilize and recruit endogenous endothelial progenitor cells (EPCs), directing their homing to ischemic regions; mechanistically, this response is associated with Akt pathway activation [51]. In rat MI models, administration of MSCs or MSC-Exos increases capillary density and perfusion, improves local hemodynamics, and translates into better global cardiac function and tissue survival [52,53].
3.2. Anti-Apoptotic
Another crucial contribution of MSCs to myocardial repair is their anti-apoptotic effect. Following MI, ischemia and hypoxia trigger extensive cardiomyocyte loss, predominantly through apoptosis, which accelerates functional deterioration. MSC therapy can attenuate this process through multiple mechanisms, limiting apoptosis induced by oxidative stress, inflammatory cytokines, and ischemic hypoxia and thereby restraining infarct expansion [54].
MSC-Exos contain microRNAs that directly modulate cardiomyocyte survival pathways. For example, miR-21-5p targets YAP1 to dampen Hippo–YAP signaling; miR-24 and miR-125b reduce the expression of pro-apoptotic mediators such as BAX and CASP3; and miR-486-5p in BM-MSC-Exos inhibits PTEN, thereby activating the pro-survival PI3K–Akt pathway [55,56,57,58]. In a murine MI model, adipose-derived MSC exosomes (AD-MSC-Exos) enriched in miR-671 suppress TGFBR2 and downstream SMAD2 phosphorylation, resulting in reduced cardiomyocyte apoptosis [59]. Mechanistically, miR-22 has been shown to bind the 3′ untranslated region of Methyl-CpG binding protein 2 (MECP2) and suppress MECP2 expression, shifting the BAX/BCL-2 balance toward survival and inhibiting CASP3 activation, thereby reducing cardiomyocyte apoptosis under ischemic conditions [60,61].
MSCs and their exosomes also mitigate apoptosis by attenuating oxidative stress. After MI, excessive reactive oxygen species (ROS) injure cardiomyocytes and trigger apoptosis. In vitro, Induced pluripotent stem cells-derived mesenchymal stromal/stem cells (iPSCs-MSCs) derived exosomes delivering miR-21 (Nanog-responsive) and miR-210 (HIF-1α responsive) suppress caspase-3/7 activation, thereby protecting cardiomyocytes against H₂O₂-induced oxidative stress and apoptosis [62]. Emerging evidence suggests that MSC-Exos can further enhance redox balance by delivering antioxidants and miRNAs like miR-1 and miR-133 to maintain mitochondrial integrity and shift metabolism toward glycolysis in hypoxic states [63].
3.3. Anti-Fibrotic
Most chronic inflammatory diseases are accompanied by fibrotic remodeling, characterized by immune cell-driven myofibroblast activation and excessive extracellular matrix (ECM) deposition, such as type I collagen. Although scar formation helps preserve structural integrity after injury, unchecked fibrosis stiffens the myocardium, impairs both systolic and diastolic function, and ultimately contributes to HF [64].
MSCs counteract cardiac fibrosis through multiple complementary mechanisms. First, MSCs mitigate fibrosis by modulating the local inflammatory milieu and promoting a pro-reparative phenotype. Specifically, through the COX-2–PGE2 pathway, MSCs drive macrophage polarization from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype, thereby decreasing the production of inflammatory cytokines such as TNF-α, IL-1β, and IL-6. MSCs also directly limit pathological ECM remodeling by reducing the excessive accumulation of collagens I and III and rebalancing matrix turnover enzymes, such as upregulating MMP-1 while normalizing TIMP-1 levels, which collectively attenuates fibrosis and prevents adverse ventricular remodeling [65,66]. Furthermore, miR-22-mediated downregulation of MECP2 may exert a dual protective effect. Beyond its anti-apoptotic role, suppression of MECP2 can relieve transcriptional repression of downstream tissue repair genes, thereby attenuating fibrotic remodeling and promoting structural recovery after injury [60]. In addition, miR-22 has been reported to interrupt TGF-β1-induced pro-fibrotic signaling in fibroblasts, as evidenced by reduced α-smooth muscle actin (α-SMA) expression (a myofibroblast marker) via the ERK/CTGF pathway [67]. Consistently, miR-22 cargo in MSC exosomes has been associated with reduced pathological scar formation and promote functional recovery in injured myocardium, underscoring the therapeutic potential of MSC-derived exosomal miR-22 in limiting post-injury fibrosis and enhancing cardiac repair [34].
4. MSC-Exos in Cardiac Repair
Building on the mechanisms outlined above, MSC-derived exosomes (MSC-Exos) represent a central paracrine messenger system through which MSCs exert many of their reparative effects. MSC-Exos are 30–150 nm extracellular vesicles containing proteins, RNAs (including mRNAs and microRNAs) and lipids. Following uptake by recipient cells, they deliver this cargo to reprogram cellular responses within the injured myocardium. Through cargo delivery to recipient cells, MSC-Exos can reprogram injury responses in the myocardium and provide a strong rationale for cell-free therapeutic development [31,32].
In experimental settings, MSC-Exos are commonly isolated by differential ultracentrifugation, density-gradient ultracentrifugation, ultrafiltration, polymer-based precipitation, or immunoaffinity capture based on membrane markers. Conditioned medium from MSC cultures is typically collected and cleared of cells, debris, and large particles by sequential low-speed centrifugation steps, followed by enrichment of exosomes using ultracentrifugation or commercial precipitation kits. To ensure purity and preserve functional integrity, preparations are typically characterized by transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), and Western blotting for canonical exosomal markers such as CD9, CD63, and CD81.
4.1. Comparison of MSC-Exos with MSC Cell Therapy
Compared with whole-cell MSC therapy, MSC-Exos can recapitulate much of their reparative activity including pro-angiogenic, anti-apoptotic, and anti-inflammatory effects, and have therefore been increasingly explored as a cell-free alternative. Beyond biological activity, MSC-Exos may offer practical advantages in safety, product consistency, and stability. From a safety perspective, MSC-Exos exhibit relatively low immunogenicity and cytotoxicity and are less likely to provoke clinically meaningful immune rejection [32]. Because they are nanoscale vesicles rather than viable cells, MSC-Exos are also less prone to causing microvascular obstruction or infusion-related inflammatory reactions; by contrast, direct MSC administration has been associated with fever, hypersensitivity, and, in some settings, pulmonary embolism [32,68]. From a manufacturing and logistics standpoint, MSC-Exos, as acellular products, do not require survival or engraftment and thus lend themselves to standardization and scalable production. They can also be engineered for enhanced targeting and may simplify storage and distribution, potentially reducing handling costs and facilitating clinical deployment [32]. Finally, the use of MSC-Exos may alleviate certain ethical and practical concerns associated with sourcing, handling, and administering viable cells.
4.2. Source-Specific Heterogeneity of MSC-Exos
The therapeutic performance of MSC-Exos may depend on their tissue of origin. The most commonly used MSC sources are bone marrow (BM-MSCs), adipose tissue (AD-MSCs), and umbilical cord (UC-MSCs). Exosomes produced by these MSCs differ in microRNA and protein cargo, which can shape their efficacy in cardiac repair. For example, BM-MSC-Exos enriched in the lncRNA MIR9-3HG mitigate I/R injury by modulating ferroptosis via the MIR9-3HG/PUM2/PRDX6 axis, thereby improving cardiac function [69]. Additionally, BM-MSC-Exos enriched in miR-210 have been reported to reduce cardiomyocyte apoptosis and infarct size and improve cardiac function after MI, potentially via an AIFM3/PI3K–AKT/p53–linked axis [70]. AD-MSC-Exos carry miR-671, which targets TGFBR2 and suppresses SMAD2 phosphorylation, reducing myocardial fibrosis and inflammation while improving cardiomyocyte survival and limiting apoptosis [59]. Similarly, AD-MSC-Exos carrying the miR-221/222 cluster attenuate myocardial I/R injury by suppressing apoptosis and hypertrophic remodeling, consistent with regulation of PUMA- and ETS-1–associated signaling [71]. UC-MSC-Exos deliver miR-19a to inhibit SOX6, activate AKT, and suppress the JNK3/caspase-3 pathway, collectively attenuating acute MI–induced injury and apoptosis [72]. Beyond the three canonical MSC sources, dental pulp MSC-Exos enriched in TSG-6 have been shown to dampen NF-κB–driven inflammation, promote macrophage polarization toward an M2 phenotype, and reduce infarct size in MI models (Table 1) [73].
From a translational standpoint, BM-MSC-Exos are the most extensively studied, but bone-marrow harvest is invasive and functional potency can be age-dependent. AD-MSC-Exos are easier to obtain and amenable to scale-up, yet donor-to-donor heterogeneity, e.g., obesity or metabolic status, may affect product consistency. UC-MSC-Exos offer practical advantages, including noninvasive tissue collection, higher expansion efficiency, and lower immunogenic and tumorigenic risk, making them attractive for standardized, large-scale clinical deployment [74].
Comparative Exosomal Cargo and Effects from Different MSC Sources in Cardiac Repair.
| MSC Source | Key Exosomal Cargo (miRNA/protein) | Main Mechanism/Pathway | Effects in Cardiac Injury | Animal Model | Reference |
| BM-MSCs | MIR9-3HG | MIR9-3HG/Pum2/PRDX6 axis | Improve in ferroptosis and I/R injury | Mouse myocardial I/R | Zhang et al. [69] |
| miR-210 | miR-210/AIFM3/PI3K–AKT/p53 axis | Improve in apoptosis, infarct size and cardiac function after MI | Rat MI | Cheng et al. [70] | |
| AD-MSCs | miR-671 | miR-671/TGFBR2/SMAD2 axis | Improve in fibrosis and inflammation, apoptosis and cardiac function after MI | Mouse MI | Wang et al. [59] |
| miR-221/222 cluster | miR-221/222/PUMA/ETS-1 axis | Improve in apoptosis, hypertrophy and myocardial I/R injury | Mouse myocardial I/R | Lai et al. [71] | |
| UC-MSCs | miR-19a | miR-19a/SOX6/AKT/JNK3/CASP3 axis | Improve in apoptosis and acute MI injury | Rat MI | Huang et al [72] |
| DP-MSCs | TSG-6 | TSG-6/NF-κB axis; M1→M2 polarization | Improve in inflammation, M2 macrophages and infarct size | Nude rat MI | Amaro-Prellezo et al. [73] |
Beyond MSC-Exos, small extracellular vesicles (sEVs) from different progenitors also display source-dependent activity. In a systematic, head-to-head comparison of sEVs from multiple cell types, embryonic stem cell–derived sEVs (ESC-sEVs) showed the most pronounced benefits across angiogenesis, anti-fibrosis, immune modulation, and improvements in cardiac function. By contrast, BM-MSC-sEVs were particularly effective at limiting early cardiomyocyte apoptosis and dampening inflammation, whereas sEVs from ESC-derived cardiomyocytes (CM-sEVs) exhibited comparatively modest overall effects [75]. These findings underscore the decisive role of vesicle provenance in cardiac repair and suggest that the choice of sEV source should be tailored to the therapeutic objective.
4.3. Prospects and Challenges
Despite encouraging therapeutic activity of MSC-Exos in preclinical models of cardiac repair, translating these findings into safe and effective interventions for routine clinical use will require overcoming several critical hurdles. These challenges span the entire bench-to-bedside pipeline, from foundational research to clinical implementation.
4.3.1. Standardized Manufacturing and Quality Control
At present, multiple approaches are used to isolate and enrich MSC-EVs, including ultracentrifugation, size-exclusion chromatography, and immunoaffinity-based capture; however, each entails trade-offs in yield, EV subpopulation specificity, scalability, and preservation of bioactivity. Consistent with MISEV2023 recommendations, the lack of harmonized manufacturing workflows, multimethod characterization, and transparent reporting standards hampers inter-study comparability and, more critically, limits robust GMP-aligned large-scale production required for clinical translation [76].
4.3.2. Heterogeneity and “Design-on-Demand” Products
The composition and function of MSC-Exos vary with MSC source, culture conditions, and even donor-to-donor differences. Accordingly, future studies should more precisely define the bioactive cargo most relevant to cardiac repair and evaluate preconditioning or genetic engineering of parent MSCs to generate exosomes with improved potency and targeting, ultimately enabling “design-on-demand” therapeutics.
4.3.3. Improving In Vivo Delivery and Targeting
Intravenous administration remains common, but the circulatory half-life, biodistribution, and myocardial accumulation of MSC-Exos are often suboptimal. Developing delivery strategies such as intramyocardial injection, hydrogel-based sustained-release systems, or surface engineering of exosomes to enhance cardiac tropism, which represents a key direction to maximize bioavailability and local efficacy while minimizing off-target effects [77].
4.3.4. Optimizing Clinical Regimens and Trial Design
Finally, defining optimal therapeutic regimens for MSC-Exos including dose, route of administration, therapeutic time window, and dosing frequency will require rigorously designed, appropriately powered clinical trials. Multicenter, randomized, controlled studies should assess both short- and long-term improvements in cardiac function and delineate the long-term safety profile, including potential immune reactions and other adverse events.
5. Preclinical Evidence of MSC Therapy in Cardiac Repair
MSCs have been extensively evaluated as a cell-based therapy for cardiac repair across diverse animal models. Efficacy has been assessed using functional, structural, and mechanistic endpoints: improvements in left ventricular ejection fraction (LVEF) by echocardiography; reductions in left ventricular end-systolic and end-diastolic volumes (LVESV, LVEDV); decreases in infarct size, increased ventricular wall thickness, and attenuation of adverse remodeling on histology; and mechanistic readouts including reduced cardiomyocyte apoptosis, increased capillary density in the infarct border zone, diminished inflammatory infiltration, and lower collagen deposition [78].
A large body of rodent studies demonstrate that MSC therapy limits infarct size, increases capillary density, and improves ejection fraction, with benefits mediated predominantly by paracrine mechanisms. Concordant findings in large-animal models with cardiac anatomy closer to humans corroborate these effects [79,80,81,82]. Collectively, these preclinical data support the therapeutic efficacy of MSCs, emphasize paracrine signaling as the principal mode of action, and provide the rationale for subsequent clinical trials.
6. Clinical Evidence and Trials of MSC Therapy in Cardiac Repair
Building on an extensive body of encouraging preclinical data, hundreds of clinical trials worldwide have been initiated to evaluate the safety and efficacy of MSC therapy for cardiac repair and related cardiac conditions (Table 2).
6.1. Phase I Trials: Establishing Safety and Feasibility
Phase I study primarily aims to establish the safety and feasibility of administering MSCs to patients with cardiac disease. These early trials typically enroll small cohorts with ischemic cardiomyopathy (ICM) or HF and focus on potential adverse events such as arrhythmias, immune reactions, and systemic infusion-related responses.
A representative example is the POSEIDON trial (Percutaneous Stem Cell Injection Delivery Effects on Neomyogenesis), which randomized 30 patients with ICM to transendocardial injection of either autologous or allogeneic BM-MSCs. Based on the 13-month follow-up of the POSEIDON trial, transendocardial injection of MSCs in patients with ICM showed favorable effects. In terms of ventricular remodeling, both autologous and allogeneic MSCs were associated with significant reductions in infarct size and left-ventricular end-diastolic volume. However, global ejection fraction did not increase significantly, suggesting that improvements in contractile function may be more regional or dose-dependent (as indicated in the lower-dose cohort). Although the two groups performed similarly on remodeling endpoints, significant gains in functional measures such as 6-min walk distance and quality-of-life scores were observed only in the autologous arm. Regarding safety, both autologous and allogeneic MSCs were well tolerated, with a low incidence of serious adverse events. Notably, allogeneic MSCs did not trigger significant immune reactions or additional serious adverse events, providing preliminary evidence consistent with low immunogenicity and potential immunomodulatory activity [28].
A Phase I study led by the Cardiovascular Cell Therapy Research Network (CCTRN) evaluated transendocardial allogeneic BM-MSC delivery in 31 cancer survivors with anthracycline-induced cardiomyopathy (AIC). Cardiac MRI was used to assess LVEF and global strain. No serious adverse events (e.g., immune reactions or de novo malignancies) were observed. While myocardial viability metrics (e.g., scar size and viable mass) did not significantly improve and LVEF changes were not statistically different (mean ΔLVEF: 3.47% vs. 2.68%; p = 0.746), quality of life (Minnesota Living with HF Questionnaire) improved significantly (p = 0.048) [83].
The TAC-HFT trial evaluated transendocardial BM-MSCs versus bone-marrow mononuclear cells (BMCs) in patients with chronic ICM. Cell delivery was safe and associated with signals of functional improvement; however, the primary endpoint the 30-day rate of treatment-related serious adverse events did not demonstrate superiority over placebo [84]. Complementing this, a 2024 Phase I trial by Kawamura et al. tested allogeneic adipose-derived MSC spray transplantation in 10 ICM patients, demonstrating procedural safety, no immune events, and preliminary angiogenesis benefits via improved perfusion imaging [30].
Selected Clinical Trials of MSC Therapy for Cardiac Repair.
| Clinical Trial Registration | Study Type | Number of Participants (MSC Group/Control Group) | Patient Population | Mean Age (MSC Group/Control Group) | Mean LVEF at Baseline (MSC Group/Control Group) | NYHA Class III and IV (%) (MSC Group/Control Group) | Method of Stem Cell Delivery | Type of MSC | Primary Endpoint | Follow-Up Time | Key Efficacy Result | Author and Year |
| NCT01087996 | Phase I/II | 15; 15 | LV dysfunction due to ICM | 62.8; 63.7 | 27.7; 26.3 | 26.7; 26.7 | Transendocardial | Allo and Auto-BM- MSCs | 30-day incidence of treatment-emergent SAEs | 13 Months | Met safety goal; improved functional capacity, QoL, and remodeling | Hare et al. (2012) [28] |
| NCT02509156 | Phase I | 14; 17 | Chronic AIC | 54.7 ± 12.8; 58.2 ± 11.2 | 33.7 ± 3.4; 32.5 ± 6.5 | 7; 24 | Transendocardial | Auto-BM- MSCs | Safety and feasibility of Allo-MSCs | 12 Months | Safe and feasible; improved QoL and 6MWD | Bolli, et al. (2020) [83] |
| NCT00768066 | Phase I/II | 19; 11 | ICM and LVEF < 50% | 57.1; 60.0 | 35.7;2 8.1 | 10.5; 30 | Transendocardial | Auto-BM- MSCs | 30-day composite SAE rate | 12 Months | Safe; improved exercise capacity, QoL, and reduced infarct size | Heldman et al. (2014) [84] |
| NCT01739777 | Phase I/II | 15; 15 | ICM and LVEF < 40% | 57.20 ± 11.64; 57.33 ± 10.05 | 31.49 ± 4.71; 33.00 ± 6.18 | - | Intravenous infusion | Allo-UC-MSCs | Safety (infusion-related AEs & immunology) | 12 Months | Safe; significantly improved LVEF, NYHA class, and QoL | Bartolucci et al. (2017) [85] |
| NCT00644410 | Phase II | 40; 20 | CMI and end-stage HF | 66.1 + 7.7; 64.2 + 10.6 | 28.2 + 9.3; 25.1 + 8.5 | 72.5; 75 | Intramyocardial | Auto-BM- MSCs | Change in LVESV at 6 months (MRI) | 6 Months | Reduced LVESV and improved LVEF; no change in NYHA/6MWD | Mathiasen et al. (2015) [86] |
| NCT02032004 | Phase III | 283; 282 | High-risk HF with HFrEF | 62.7 ± 10.9; 62.6 ± 10.4 | 28.6 ± 6.7; 28.6 ± 6.9 | 61.8; 63.1 | Transendocardial | Auto-MPS | Time-to-first recurrent HF event | 12 Months | Neutral primary outcome; reduced MI/stroke risk in subgroups | Perin et al. (2023) [87] |
6.2. Phase II Trials: Efficacy Signals and Dose-Finding
Building on established safety, Phase II trials begin to explore preliminary efficacy, typically incorporating control groups and using changes in LVEF, New York Heart Association (NYHA) class, and quality-of-life scores as primary or key secondary endpoints.
In RIMECARD, UC-MSCs were favored for their “off-the-shelf” availability. In patients with stable chronic heart failure with reduced ejection fraction (HFrEF), a single intravenous dose of UC-MSCs at 1 × 106 cells/kg was safe and feasible, and yielded a significantly greater 12-month improvement in LVEF compared with controls (+7.07% vs +1.85%; p = 0.028). NYHA class and quality-of-life measures also improved, with no effect on mortality or rehospitalization [85].
The MSC-HF trial evaluated intramyocardial injection of autologous BM-MSCs in ischemic HF. At 6-month follow-up, the procedure was well tolerated and produced significant improvements in left-ventricular structure and function—including LVESV, LVEF, stroke volume (SV), and LV mass—whereas functional status and quality-of-life measures (NYHA class, 6-min walk test, and KCCQ) did not show significant changes [86]. Beyond ischaemic HF, the 2024 SCIENCE II pilot extended CSCC-ASC evaluation to non-ischaemic HFrEF, using LVESV as the primary endpoint and reporting signals of improved LV remodelling and functional status, highlighting that underlying HF aetiology and patient phenotype may influence responsiveness [88].
6.3. Phase III Trials: Confirmatory Testing and Clinical Outcomes
Upon advancing to Phase III, which is designed to generate confirmatory evidence for regulatory approval MSC therapy has faced greater challenges, including larger patient cohorts, stricter endpoints, and the need to demonstrate consistency of effect. In DREAM-HF, 565 high-risk patients with HFrEF received a single transendocardial injection of mesenchymal precursor cells (MPCs, an MSC subpopulation). The trial did not meet its primary composite endpoint of major cardiac events (heart-failure hospitalization or resuscitated ventricular arrhythmia). However, the treatment arm showed a 58% reduction in nonfatal MI and stroke versus control (HR 0.42; 95% CI 0.23–0.76), with even greater benefit in the inflammatory subgroup defined by baseline hsCRP ≥ 2 mg/L (75% risk reduction; HR 0.25; 95% CI 0.09–0.66) [87]. More recently, an inflammation-focused analysis of DREAM-HF further supported a biomarker-stratified view: baseline hsCRP ≥2 mg/L and ischaemic aetiology identified higher-risk control patients, whereas a single intramyocardial MPC administration produced the most pronounced and sustained reductions in 2-point (MI or stroke) and 3-point (CV death/MI/stroke) MACE in the ischaemic HFrEF + inflammation subgroup. These data suggest that MSC/MPC benefit may be “phenotype-dependent”, shifting the trial logic from expecting uniform reverse remodelling toward targeting inflammatory risk and event-driven outcomes in biomarker-enriched populations [89].
In contrast, the Phase IIb/III SCIENCE trial (2023) offered a more cautionary perspective [90]. This multicenter study evaluated allogeneic adipose-derived MSCs (CSCC-ASC) in patients with ischemic HFrEF. Unlike the event-reduction signals seen in DREAM-HF, the SCIENCE trial failed to meet its primary endpoint of reducing LVESV or improving functional capacity at 12 months compared to placebo. This divergence highlights a critical controversy in the field: the inconsistency potentially stemming from cell sources and the inadequacy of traditional structural endpoints to capture the immunomodulatory benefits of MSCs. Importantly, a 2025 pooled analysis combining the SCIENCE trial with a Danish ASC study (total n = 214) similarly found that a single intramyocardial CSCC-ASC injection in chronic ischaemic no-option HF did not improve cardiac function, exercise capacity, or quality of life at 6 months, reinforcing the challenge of achieving consistent efficacy in advanced ischaemic phenotypes [91].
Additional evidence comes from a recent meta-analysis. In patients with acute MI, MSC therapy has generally shown an acceptable safety profile and modest improvement in left ventricular function, particularly within the first 6–12 months after treatment. However, current pooled clinical evidence has not consistently demonstrated reductions in hard clinical endpoints such as MACE, HF hospitalization, or cardiovascular mortality [92]. Therefore, although MSC therapy remains promising as an adjunctive strategy after acute MI, its ability to prevent post-MI HF requires confirmation in rigorously designed, adequately powered trials with reliable long-term clinical endpoints.
To date, no MSC product has received FDA or EMA approval for routine cardiac indications. Although recent late-stage trials and pooled analyses continue to support an acceptable safety profile, the lack of consistent success in meeting primary structural endpoints suggests that future pivotal trials may need to pivot away from “regeneration” (pumping function) toward “event reduction” and inflammatory modulation as primary targets for clinical translation.
6.4. Safety, Limitations, and Controversies in MSC Cardiac Therapy
In summary, although numerous trials have investigated MSC therapy for cardiac repair, outcomes have been inconsistent. Most studies confirm that MSC therapy is safe but demonstrate only modest improvements in cardiac function or patient outcomes. A primary biological barrier is the low retention and engraftment rate of transplanted cells. Transplanted MSCs largely wash out or perish shortly after administration, leading to low myocardial retention and transient benefits [93,94]. Additionally, the field faces practical challenges, including complex regulatory and manufacturing hurdles and high treatment costs, which hinder widespread clinical adoption [95]. High-profile trials like the phase III DREAM-HF exemplify these issues by confirming excellent safety but failing to significantly improve primary heart failure endpoints, although certain subgroups showed hints of benefit [87]. Furthermore, this field must navigate skepticism arising from historical “cell therapy fraud” debates, which forces current MSC studies to establish more stringent validation standards [96]. Nevertheless, adaptive strategies are under active investigation to enhance outcomes. For example, cell-free approaches utilizing MSC-derived exosomes aim to circumvent retention issues, and improved delivery techniques are being developed to bolster efficacy, offering a realistic path for iterative improvement. Recent meta-analyses updated through 2023–2024 continue to support an overall favourable safety profile and modest improvements in functional and imaging indices (e.g., LVEF), while underscoring substantial heterogeneity across cell sources, delivery routes, and endpoints—consistent with the mixed signals observed in contemporary Phase III programmes [97].
6.5. Clinical Trial Design and Outcome Synthesis
Clinical studies suggest that a patient’s inflammatory status is a crucial determinant of the efficacy of MSC therapy. A subgroup analysis of the DREAM-HF study found that in patients with high inflammation and baseline high-sensitivity C-reactive protein (hsCRP ≥ 2 mg/L), BM-MSC therapy significantly reduced the risk of non-fatal myocardial infarction or stroke and the risk of major cardiovascular events [98]. Johnston et al. also pointed out that a significant decrease in major adverse cardiovascular events was only observed in patients with evidence of “maladaptive inflammation” in DREAM-HF, suggesting that MSCs play a key role in regulating inflammation [99]. Conversely, in patients without significant systemic inflammation, the improvement in systolic function by MSCs is often weak; for example, in an anthracycline-induced cardiomyopathy study, MSCs only improved quality of life scores without significantly increasing LVEF [83].
Cell origin also affects therapeutic efficacy. BM-MSCs have been studied the most and are considered to have strong differentiation and immunomodulatory potential, in contrast, AD-MSCs are easier to obtain and have higher yields, however, recent large-scale trials have not shown clear benefits. UC-MSCs grow rapidly and have low immunogenicity, making them suitable for allogeneic use. For example, in the RIMECARD trial, intravenous injection of UC-MSCs significantly increased LVEF from baseline (+7.1%, p = 0.028), improving NYHA classification and quality of life [98,100]. Overall, different MSC sources have their own advantages in terms of cell function and practicality, but there is still a lack of direct head-to-head clinical comparisons, suggesting that more research is needed to clarify which source is most effective in specific disease contexts.
The route of administration and timing of MSC administration also affect efficacy. For example, direct intracardiac injection (such as POSEIDON, TAC-HFT) can deliver high concentrations of cells to the target area, but the procedure is technically complex and less easily scalable. Intravenous infusion is convenient but may be limited by lower myocardial retention and first-pass pulmonary trapping [80]. Timing is another key variable: meta-analyses suggest that MSC delivery within the early post-AMI window, particularly within the first week or within 2–14 days after PCI, may be associated with greater improvement in LVEF[101,102]. However, evidence for durable reductions in HF events remains insufficient. In contrast, chronic HF may require repeated, higher-dose, or more targeted delivery strategies to sustain therapeutic effects. These factors lead to differences in efficacy in different trials and should be weighed and explored in trial design.
Trials using traditional ventricular structural/functional indicators such as LVEF and ventricular volume, as primary endpoints often fail to capture the immunomodulatory effects of MSCs. For example, the SCIENCE trial, which used a reduction in LVESV as the endpoint, showed no difference. In contrast, the DREAM-HF trial used events such as heart failure hospitalization and fatal arrhythmias as the primary composite endpoint [90,99]. Although this composite endpoint was not significant overall, its subgroup analysis highlighted the ability of MSCs to significantly reduce events such as myocardial infarction/stroke in patients with high inflammation. This shift in focus from structural restoration to event reduction represents a new research paradigm. As the commentary notes, the DREAM-HF results suggest that we should view MSCs as an anti-inflammatory “immunotherapy” rather than simply a regenerative therapy, and use the reduction of adverse cardiovascular events as a key criterion for evaluating efficacy.
7. iPSC-MSCs: A Next-Generation Cell Source for Cardiac Therapy
iPSCs-MSCs are produced by reprogramming somatic cells (e.g., fibroblasts) into iPSCs followed by directed differentiation into MSC-like progeny. Compared with traditional tissue-derived MSCs, iPSCs-MSCs offer potential advantages in expandability, batch-to-batch consistency, and a “rejuvenated”/de-senescent phenotype. These features can mitigate donor-to-donor variability and the functional drift and replicative senescence that accrue with extended passaging, thereby facilitating standardized, large-scale manufacturing for clinical translation [103,104].
In rodent models of myocardial I/R injury and chronic ICM, iPSCs-MSCs improve cardiac structure and function via pro-angiogenic, anti-fibrotic, and anti-apoptotic mechanisms. In a controlled, head-to-head study, intramyocardial delivery of iPSCs-MSCs in a rat I/R model produced a greater increase in LVEF than either BM-MSCs or vehicle control [105]. Additionally, iPSCs-MSC–derived exosomes (iPSCs-MSC-Exos) carrying microRNAs (e.g., miR-202-5p) have been shown to inhibit cardiomyocyte pyroptosis and enhance cardiac function [106]. Collectively, these findings suggest that iPSCs-MSCs and their exosomes may offer superior therapeutic potential over conventional MSCs for cardiac repair, warranting further translational investigation.
Early human data in non-cardiovascular indications provide important safety signals for iPSCs-MSCs. In a multicenter Phase I study of CYP-001 (Cymerus® iPSC-MSCs; Cynata Therapeutics Limited, Carlton, VIC, Australia; manufactured by Waisman Biomanufacturing, University of Wisconsin–Madison, Madison, WI, USA) for steroid-refractory acute graft-versus-host disease (SR-aGVHD), two intravenous infusions were safe and well tolerated; the 100-day overall response rate (ORR) and complete response rate (CRR) were 86.7% and 53.3%, respectively. Over two years of follow-up, no treatment-related serious adverse events, malignancies, or other safety concerns were observed [107]. These findings support a favorable safety profile for iPSC-MSCs and provide a foundation for their clinical translation to other indications, including CVD.
The principal safety risk of iPSCs-MSCs therapy is tumorigenicity specifically, teratoma formation arising from residual undifferentiated pluripotent stem cells (PSCs). Consequently, highly sensitive detection and clearance of residual PSCs constitute a critical quality-control checkpoint for iPSCs-MSCs products. Mitigation requires validated assays capable of detecting rare residual PSCs including molecular, flow-cytometric, and functional readouts GLP-compliant tumorigenicity studies, and risk-reduction strategies such as gene-engineered “safety switches” (e.g., inducible suicide systems) or selective pharmacologic ablation. Recent position papers and methodological studies provide a framework for internationally harmonized evaluation procedures [108,109].
8. MSC in Cardiac Repair: Conclusion and Future Directions
Over several decades, the field has shifted from an early “cell-replacement” notion of MSC therapy to a paracrine-centric paradigm that now confronts complex translational challenges. This review has systematically summarized the mechanisms of action of MSCs, iPSCs-MSCs and their exosomes in cardiac repair, tracing the trajectory from preclinical studies to clinical trials. Taken together, MSC-based therapies have made notable progress. Across preclinical and clinical investigations they have consistently demonstrated a favorable safety profile, immunomodulatory potential, and modest improvements in ventricular function [92]. However, widespread clinical adoption remains constrained by several barriers: extremely low engraftment and cell survival in the ischemic myocardium, heterogeneity in MSC sources and manufacturing leading to inconsistent efficacy, and regulatory concerns about long-term safety.
Rather than representing endpoints, these limitations define clear priorities for the next phase of innovation. Future directions in MSC-based cardiac repair can be conceptually organized into three interrelated thematic clusters—product optimization, delivery and tracking, and clinical translation—which together outline a coherent and realistic roadmap from bench to bedside.
8.1. Product Optimization: Toward Safer, More Potent, and Standardized Therapeutics
The first major priority centers on optimizing the therapeutic product itself. A key trend is the transition from cell-based to cell-free platforms, with MSC-derived exosomes emerging as next-generation therapeutics. As acellular nanoscale vesicles, exosomes substantially mitigate risks inherent to live cell transplantation, including immune rejection, microvascular obstruction, and tumorigenicity associated with engrafted cells. Their ability to traverse biological barriers and efficiently deliver bioactive cargos—such as proteins, mRNAs, and miRNAs—to injured myocardium further enhances their therapeutic appeal. In addition, exosomes offer practical advantages in storage, transport, and dose standardization, as they can be mass-produced and stored long-term without significant loss of potency [63].
Concurrently, next-generation cell sources, particularly iPSC-derived MSCs (iPSC-MSCs), address intrinsic limitations of tissue-derived MSCs, such as donor-to-donor variability, limited expandability, and replicative senescence. iPSC-MSCs provide a virtually unlimited and more homogeneous cell supply, laying a foundation for standardized, high-quality manufacturing [110]. However, rigorous quality control will be essential to eliminate residual undifferentiated iPSCs and minimize tumorigenicity risk, with long-term validation in large-animal models and clinical studies remaining a critical requirement.
Finally, potency enhancement through genetic engineering and preconditioning represents an important complementary strategy. By overexpressing survival pathways (e.g., Akt) or homing receptors (e.g., CXCR4), MSCs can be engineered to better withstand the hostile ischemic microenvironment and more effectively target sites of myocardial injury [111]. Notably, these optimization strategies are not mutually exclusive and may converge, for example, through the development of designer exosomes carrying defined cardioprotective cargos to achieve reproducible and scalable potency beyond natural biological variance.
8.2. Delivery and Tracking: Improving Retention, Targeting, and Functional Integration
The second thematic cluster focuses on delivery efficiency and in vivo tracking, which remain major bottlenecks for both cell- and exosome-based therapies. Biomaterial integration, particularly the use of injectable or implantable hydrogels, has emerged as a promising approach to enhance retention and survival within the infarcted myocardium. Of particular interest are conductive hydrogels, which not only improve mechanical anchoring of transplanted cells or vesicles but also facilitate electrical coupling with host myocardium, potentially reducing arrhythmogenic risk [112]. In parallel, advances in targeting strategies and imaging technologies are increasingly important. Surface modification of exosomes or MSCs to enhance myocardial homing, combined with non-invasive imaging modalities for real-time tracking, will be essential to better understand biodistribution, persistence, and mechanism of action in vivo. These delivery and tracking innovations are tightly linked to product optimization, as therapeutic efficacy ultimately depends on the ability of optimized products to reach and act within the injured myocardium.
8.3. Clinical Translation: Precision Medicine, Trial Design, and Regulatory Pathways
The third and final cluster addresses clinical translation, where the gap between biological promise and measurable patient benefit remains most evident. Increasing evidence from late-stage trials and meta-analyses highlights that, while MSC therapies are generally safe, they have not yet consistently reduced hard clinical endpoints such as long-term heart failure hospitalization or mortality. This underscores the need for precision medicine approaches, including biomarker-guided patient stratification and AI-enabled genomic profiling, to identify subgroups most likely to benefit from MSC-based interventions.
Equally important is the evolution of clinical trial design and endpoints. Beyond traditional measures of left ventricular ejection fraction, future trials may need to incorporate composite or mechanistic endpoints—such as inflammation resolution, myocardial energetics, or fibrosis modulation—to more accurately capture therapeutic impact. In parallel, navigating regulatory frameworks, particularly for exosome-based and engineered products, will require early engagement with regulatory agencies to define standards for manufacturing, quality control, and long-term safety assessment.
Importantly, these three clusters are deeply interconnected. Product optimization strategies directly influence delivery efficiency, while advances in delivery and tracking inform patient selection and trial design. In terms of translational timelines, incremental improvements in delivery systems and biomarker-guided stratification are likely achievable in the near term, whereas fully standardized exosome therapeutics and large-scale clinical deployment of iPSC-MSCs may represent mid- to long-term goals. Together, these converging efforts define a realistic pathway toward overcoming current barriers. In conclusion, although the path to widespread clinical adoption of MSC-based cardiac repair remains challenging, it is increasingly well-defined. By systematically advancing product optimization, refining delivery and tracking strategies, and aligning clinical translation with precision medicine principles, multidisciplinary innovation can unlock the full therapeutic potential of MSCs and their derivatives. Such progress will be essential to ultimately deliver meaningful and durable benefits to the growing population of patients suffering from cardiovascular disease worldwide.
Author Contributions
Z.W. and B.G.: conceived the review; Z.W.: writing—original draft preparation, made the figures; J.Z.: software; N.G.: reviewed the manuscript; B.G.: supervision, project administration, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Use of AI and AI-Assisted Technologies
During the preparation of this work, the author used ChatGPT (OpenAI) to search literature and edit language, including improvements to grammar, clarity, and readability. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
References
- 1.
Vos, T.; Lim, S.S.; Abbafati, C.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- 2.
Mensah, G.A.; Fuster, V.; Murray, C.J.; et al. Global burden of cardiovascular diseases and risks, 1990–202 J. Am. Coll. Cardiol. 2023, 82, 2350–2473.
- 3.
Chong, B.; Jayabaskaran, J.; Jauhari, S.M.; et al. Global burden of cardiovascular diseases: Projections from 2025 to 2050. Eur. J. Prev. Cardiol. 2025, 32, 1001–1015.
- 4.
Martin, S.S.; Aday, A.W.; Allen, N.B.; et al. 2025 heart disease and stroke statistics: A report of US and global data from the American Heart Association. Circulation 2025, 151, e41–e660.
- 5.
McDonagh, T.A.; Metra, M.; Adamo, M.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599.
- 6.
Timmis, A.; Vardas, P.; Townsend, N.; et al. European Society of Cardiology: Cardiovascular disease statistics 2021. Eur. Heart J. 2022, 43, 716–799.
- 7.
Derks, W.; Rode, J.; Collin, S.; et al. A latent cardiomyocyte regeneration potential in human heart disease. Circulation 2025, 151, 245–256.
- 8.
Feng, L.; Su, L.; Ren, J. Cardiac contractility modulation to enhance optimized medical therapy and improve cardiac remodeling in advanced heart failure: A case report. Front. Cardiovasc. Med. 2025, 12, 1577680.
- 9.
Al Hazzouri, A.; Attieh, P.; Sleiman, C.; et al. Left ventricular assist device in advanced refractory heart failure: A comprehensive review of patient selection, surgical approaches, complications and future perspectives. Diagnostics 2024, 14, 2480.
- 10.
Piotrowska, P.; Kraskiewicz, H.; Klimczak, A. Mesenchymal Stem Cell Secretome for Cardiac Regeneration: Opportunity for Cell-Free Therapy. Int. J. Mol. Sci. 2025, 27, 209.
- 11.
Rofaani, E.; Mardani, M.; Yutiana, P.; et al. Differentiation of mesenchymal stem cells into vascular endothelial cells in 3D culture: A mini review. Mol. Biol. Rep. 2024, 51, 781.
- 12.
Szaraz, P.; Gratch, Y.S.; Iqbal, F.; et al. In vitro differentiation of human mesenchymal stem cells into functional cardiomyocyte-like cells. J. Vis. Exp. JoVE 2017, 126, 55757.
- 13.
Friedenstein, A.; Chailakhjan, R.; Lalykina, K. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Prolif. 1970, 3, 393–403.
- 14.
Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650.
- 15.
Wilson, A.; Hodgson-Garms, M.; Frith, J.E.; et al. Multiplicity of mesenchymal stromal cells: Finding the right route to therapy. Front. Immunol. 2019, 10, 1112.
- 16.
Margiana, R.; Markov, A.; Zekiy, A.O.; et al. Clinical application of mesenchymal stem cell in regenerative medicine: A narrative review. Stem Cell Res. Ther. 2022, 13, 366.
- 17.
Teoh, P.L.; Akhir, H.M.; Ajak, W.A.; et al. Human mesenchymal stromal cells derived from perinatal tissues: Sources, characteristics and isolation methods. Malays. J. Med. Sci. 2023, 30, 55.
- 18.
Kostecka, A.; Kalamon, N.; Skoniecka, A.; et al. Adipose-derived mesenchymal stromal cells in clinical trials: Insights from single-cell studies. Life Sci. 2024, 351, 122761.
- 19.
Rodríguez-Fuentes, D.E.; Fernández-Garza, L.E.; Samia-Meza, J.A.; et al. Mesenchymal stem cells current clinical applications: A systematic review. Arch. Med. Res. 2021, 52, 93–101.
- 20.
Doherty, D.F.; Roets, L.; Krasnodembskaya, A.D. The role of lung resident mesenchymal stromal cells in the pathogenesis and repair of chronic lung disease. Stem Cells 2023, 41, 431–443.
- 21.
Maraldi, T.; Russo, V. Amniotic fluid and placental membranes as sources of stem cells: Progress and challenges 2.0. Int. J. Mol. Sci. 2023, 24, 16020.
- 22.
Han, Y.; Yang, J.; Fang, J.; et al. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduct. Target. Ther. 2022, 7, 92.
- 23.
Yang, G.; Fan, X.; Liu, Y.; et al. Immunomodulatory mechanisms and therapeutic potential of mesenchymal stem cells. Stem Cell Rev. Rep. 2023, 19, 1214–1231.
- 24.
Alvites, R.; Branquinho, M.; Sousa, A.C.; et al. Mesenchymal stem/stromal cells and their paracrine activity—Immunomodulation mechanisms and how to influence the therapeutic potential. Pharmaceutics 2022, 14, 381.
- 25.
Nguyen-Truong, M.; Hematti, P.; Wang, Z. Current status of myocardial restoration via the paracrine function of mesenchymal stromal cells. Am. J. Physiol.-Heart Circ. Physiol. 2021, 321, H112–H127.
- 26.
Li, P.; Ou, Q.; Shi, S.; et al. Immunomodulatory properties of mesenchymal stem cells/dental stem cells and their therapeutic applications. Cell. Mol. Immunol. 2023, 20, 558–569.
- 27.
Dominici, M.; Le Blanc, K.; Mueller, I.; et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317.
- 28.
Hare, J.M.; Fishman, J.E.; Gerstenblith, G.; et al. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: The POSEIDON randomized trial. JAMA 2012, 308, 2369–2379.
- 29.
Razeghian-Jahromi, I.; Matta, A.G.; Canitrot, R.; et al. Surfing the clinical trials of mesenchymal stem cell therapy in ischemic cardiomyopathy. Stem Cell Res. Ther. 2021, 12, 361.
- 30.
Kawamura, T.; Yoshioka, D.; Kawamura, A.; et al. Safety and therapeutic potential of allogeneic adipose-derived stem cell spray transplantation in ischemic cardiomyopathy: A phase I clinical trial. J. Transl. Med. 2024, 22, 1091.
- 31.
Tan, F.; Li, X.; Wang, Z.; et al. Clinical applications of stem cell-derived exosomes. Signal Transduct. Target. Ther. 2024, 9, 17.
- 32.
Lotfy, A.; AboQuella, N.M.; Wang, H. Mesenchymal stromal/stem cell (MSC)-derived exosomes in clinical trials. Stem Cell Res. Ther. 2023, 14, 66.
- 33.
Kundu, D.; Shin, S.Y.; Chilian, W.M.; et al. The potential of mesenchymal stem Cell-Derived exosomes in cardiac repair. Int. J. Mol. Sci. 2024, 25, 13494.
- 34.
Rayat Pisheh, H.; Sani, M. Mesenchymal stem cells derived exosomes: A new era in cardiac regeneration. Stem Cell Res. Ther. 2025, 16, 16.
- 35.
Zhu, W.; Wang, Q.; Zhang, J.; et al. Exosomes derived from mir-214-3p overexpressing mesenchymal stem cells promote myocardial repair. Biomater. Res. 2023, 27, 77.
- 36.
Pu, Y.; Li, C.; Qi, X.; et al. Extracellular vesicles from NMN preconditioned mesenchymal stem cells ameliorated myocardial infarction via miR-210-3p promoted angiogenesis. Stem Cell Rev. Rep. 2023, 19, 1051–1066.
- 37.
Mao, C.-Y.; Zhang, T.-T.; Li, D.-J.; et al. Extracellular vesicles from hypoxia-preconditioned mesenchymal stem cells alleviates myocardial injury by targeting thioredoxin-interacting protein-mediated hypoxia-inducible factor-1α pathway. World J. Stem Cells 2022, 14, 183.
- 38.
Zhu, D.; Liu, S.; Huang, K.; et al. Intrapericardial exosome therapy dampens cardiac injury via activating Foxo3. Circ. Res. 2022, 131, e135–e150.
- 39.
Yuan, J.; Yang, H.; Liu, C.; et al. Microneedle patch loaded with exosomes containing microRNA-29b prevents cardiac fibrosis after myocardial infarction. Adv. Healthc. Mater. 2023, 12, 2202959.
- 40.
Sahoo, S.; Losordo, D.W. Exosomes and cardiac repair after myocardial infarction. Circ. Res. 2014, 114, 333–344.
- 41.
Mao, L.; Yue, Z.; Zhu, D.; et al. Exosomes: Bridge metabolic regulation in cardiac repair. NPJ Biomed. Innov. 2025, 2, 21.
- 42.
Xu, C.M.; Sabe, S.A.; Brinck-Teixeira, R.; et al. Visualization of cardiac uptake of bone marrow mesenchymal stem cell-derived extracellular vesicles after intramyocardial or intravenous injection in murine myocardial infarction. Physiol. Rep. 2023, 11, e15568.
- 43.
Khan, K.; Caron, C.; Mahmoud, I.; et al. Extracellular vesicles as a cell-free therapy for cardiac repair: A systematic review and meta-analysis of randomized controlled preclinical trials in animal myocardial infarction models. Stem Cell Rev. Rep. 2022, 18, 1143–1167.
- 44.
Jin, X.; Liu, Z. Multifaceted mechanisms and targeted delivery of mesenchymal stem cell-derived exosomes in cardiovascular diseases: A translational medicine perspective. Front. Med. 2025, 12, 1729698.
- 45.
Hegde, M.; Singh, A.K.; Kannan, S.; et al. Therapeutic applications of engineered mesenchymal stromal cells for enhanced angiogenesis in cardiac and cerebral ischemia. Stem Cell Rev. Rep. 2024, 20, 2138–2154.
- 46.
Chu, Q.; Jiang, X.; Xiao, Y. Rebuilding the myocardial microenvironment to enhance mesenchymal stem cells-mediated regeneration in ischemic heart disease. Front. Bioeng. Biotechnol. 2024, 12, 1468833.
- 47.
Li, Q.; Hou, H.; Li, M.; et al. CD73+ mesenchymal stem cells ameliorate myocardial infarction by promoting angiogenesis. Front. Cell Dev. Biol. 2021, 9, 637239.
- 48.
Tang, X.-L.; Wysoczynski, M.; Gumpert, A.M.; et al. Intravenous infusions of mesenchymal stromal cells have cumulative beneficial effects in a porcine model of chronic ischaemic cardiomyopathy. Cardiovasc. Res. 2024, 120, 1939–1952.
- 49.
Li, K.; Luo, R.; Yu, X.; et al. Enhanced human adipose-derived stem cells with VEGFA and bFGF mRNA promote stable vascular regeneration and improve cardiac function following myocardial infarction. Clin. Transl. Med. 2025, 15, e70250.
- 50.
Pan, Q.; Wang, Y.; Lan, Q.; et al. Exosomes derived from mesenchymal stem cells ameliorate hypoxia/reoxygenation-injured ECs via transferring MicroRNA-126. Stem Cells Int. 2019, 2019, 2831756.
- 51.
Deng, Q.; Du, F.; Pan, S.; et al. Activation of angiopoietin-1 signaling with engineering mesenchymal stem cells promoted efficient angiogenesis in diabetic wound healing. Stem Cell Res. Ther. 2025, 16, 75.
- 52.
Charles, C.J.; Li, R.R.; Yeung, T.; et al. Systemic mesenchymal stem cell-derived exosomes reduce myocardial infarct size: Characterization with MRI in a porcine model. Front. Cardiovasc. Med. 2020, 7, 601990.
- 53.
Zhang, Z.; Yang, J.; Yan, W.; et al. Pretreatment of cardiac stem cells with exosomes derived from mesenchymal stem cells enhances myocardial repair. J. Am. Heart Assoc. 2016, 5, e002856.
- 54.
Khubutiya, M.S.; Vagabov, A.V.; Temnov, A.A.; et al. Paracrine mechanisms of proliferative, anti-apoptotic and anti-inflammatory effects of mesenchymal stromal cells in models of acute organ injury. Cytotherapy 2014, 16, 579–585.
- 55.
Ji, Z.; Wang, C. Mesenchymal stem cell-derived exosomal mir-21-5p inhibits YAP1 expression and improves outcomes in myocardial infarction. BMC Cardiovasc. Disord. 2024, 24, 547.
- 56.
Zhang, C.; Shao, K.; Liu, C.; et al. Hypoxic preconditioning BMSCs-exosomes inhibit cardiomyocyte apoptosis after acute myocardial infarction by upregulating microRNA-24. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6691–6699.
- 57.
Zhang, B.; Mao, S.; Liu, X.; et al. MiR-125b inhibits cardiomyocyte apoptosis by targeting BAK1 in heart failure. Mol. Med. 2021, 27, 72.
- 58.
Sun, X.-H.; Wang, X.; Zhang, Y.; et al. Exosomes of bone-marrow stromal cells inhibit cardiomyocyte apoptosis under ischemic and hypoxic conditions via miR-486-5p targeting the PTEN/PI3K/AKT signaling pathway. Thromb. Res. 2019, 177, 23–32.
- 59.
Wang, X.; Zhu, Y.; Wu, C.; et al. Adipose-derived mesenchymal stem cells-derived exosomes carry microRNA-671 to alleviate myocardial infarction through inactivating the TGFBR2/Smad2 axis. Inflammation 2021, 44, 1815–1830.
- 60.
Feng, Y.; Huang, W.; Wani, M.; et al. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS ONE 2014, 9, e88685.
- 61.
Wang, Y.; Wang, H.; Tan, J.; et al. Therapeutic effect of mesenchymal stem cells and their derived exosomes in diseases. Mol. Biomed. 2025, 6, 34.
- 62.
Wang, Y.; Zhang, L.; Li, Y.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69.
- 63.
Chae, C.-W.; Choi, G.; Yoon, T.; et al. Exosome-based therapy in cardiovascular diseases: A new frontier in cardiovascular disease treatment. Korean Circ. J. 2025, 55, 461–480.
- 64.
Rockel, J.S.; Rabani, R.; Viswanathan, S. Anti-fibrotic mechanisms of exogenously-expanded mesenchymal stromal cells for fibrotic diseases. In Seminars in cell & developmental biology, 2020; Elsevier: Vol. 101, pp 87–103.
- 65.
Jin, L.; Deng, Z.; Zhang, J.; et al. Mesenchymal stem cells promote type 2 macrophage polarization to ameliorate the myocardial injury caused by diabetic cardiomyopathy. J. Transl. Med. 2019, 17, 251.
- 66.
Guo, J.; Lin, G.-s.; Bao, C.-y.; et al. Anti-inflammation role for mesenchymal stem cells transplantation in myocardial infarction. Inflammation 2007, 30, 97–104.
- 67.
Kuse, N.; Kamio, K.; Azuma, A.; et al. Exosome-derived microRNA-22 ameliorates pulmonary fibrosis by regulating fibroblast-to-myofibroblast differentiation in vitro and in vivo. J. Nippon Med. Sch. 2020, 87, 118–128.
- 68.
Nikfarjam, S.; Rezaie, J.; Zolbanin, N.M.; et al. Mesenchymal stem cell derived-exosomes: A modern approach in translational medicine. J. Transl. Med. 2020, 18, 449.
- 69.
Zhang, J.-K.; Zhang, Z.; Guo, Z.-A.; et al. The BMSC-derived exosomal lncRNA Mir9-3hg suppresses cardiomyocyte ferroptosis in ischemia-reperfusion mice via the Pum2/PRDX6 axis. Nutrition, Metabolism and Cardiovascular Diseases 2022, 32, 515–527.
- 70.
Cheng, H.; Chang, S.; Xu, R.; et al. Hypoxia-challenged MSC-derived exosomes deliver miR-210 to attenuate post-infarction cardiac apoptosis. Stem Cell Res. Ther. 2020, 11, 224.
- 71.
Lai, T.-C.; Lee, T.-L.; Chang, Y.-C.; et al. MicroRNA-221/222 mediates ADSC-exosome-induced cardioprotection against ischemia/reperfusion by targeting PUMA and ETS-1. Front. Cell Dev. Biol. 2020, 8, 569150.
- 72.
Huang, L.; Yang, L.; Ding, Y.; et al. Human umbilical cord mesenchymal stem cells-derived exosomes transfers microRNA-19a to protect cardiomyocytes from acute myocardial infarction by targeting SOX6. Cell Cycle 2020, 19, 339–353.
- 73.
Amaro-Prellezo, E.; Gómez-Ferrer, M.; Hakobyan, L.; et al. Extracellular vesicles from dental pulp mesenchymal stem cells modulate macrophage phenotype during acute and chronic cardiac inflammation in athymic nude rats with myocardial infarction. Inflamm. Regen. 2024, 44, 25.
- 74.
Li, Y.-L.; Chen, E.-G.; Ren, B.-B. Umbilical cord-derived mesenchymal stromal cells: Promising therapy for heart failure. World J. Cardiol. 2025, 17, 101153.
- 75.
González-King, H.; Rodrigues, P.G.; Albery, T.; et al. Head-to-head comparison of relevant cell sources of small extracellular vesicles for cardiac repair: Superiority of embryonic stem cells. J. Extracell. Vesicles 2024, 13, e12445.
- 76.
Welsh, J.A.; Goberdhan, D.C.; O'Driscoll, L.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404.
- 77.
Choi, H.; Choi, Y.; Yim, H.Y.; et al. Biodistribution of exosomes and engineering strategies for targeted delivery of therapeutic exosomes. Tissue Eng. Regen. Med. 2021, 18, 499–511.
- 78.
Poomani, M.S.; Mariappan, I.; Perumal, R.; et al. Mesenchymal stem cell (MSCs) therapy for ischemic heart disease: A promising frontier. Glob. Heart 2022, 17, 19.
- 79.
Mu, Y.; Cao, G.; Zeng, Q.; et al. Transplantation of induced bone marrow mesenchymal stem cells improves the cardiac function of rabbits with dilated cardiomyopathy via upregulation of vascular endothelial growth factor and its receptors. Exp. Biol. Med. 2011, 236, 1100–1107.
- 80.
Kanelidis, A.J.; Premer, C.; Lopez, J.; et al. Route of delivery modulates the efficacy of mesenchymal stem cell therapy for myocardial infarction: A meta-analysis of preclinical studies and clinical trials. Circ. Res. 2017, 120, 1139–1150.
- 81.
Qi, C.; Ma, G.; Liu, N.; et al. Transplantation of magnetically labeled mesenchymal stem cells improves cardiac function in a swine myocardial infarction model. Chin. Med. J. 2008, 121, 544–550.
- 82.
Lim, M.; Wang, W.; Liang, L.; et al. Intravenous injection of allogeneic umbilical cord-derived multipotent mesenchymal stromal cells reduces the infarct area and ameliorates cardiac function in a porcine model of acute myocardial infarction. Stem Cell Res. Ther. 2018, 9, 129.
- 83.
Bolli, R.; Perin, E.C.; Willerson, J.T.; et al. Allogeneic mesenchymal cell therapy in anthracycline-induced cardiomyopathy heart failure patients: The CCTRN SENECA trial. Cardio Oncol. 2020, 2, 581–595.
- 84.
Heldman, A.W.; DiFede, D.L.; Fishman, J.E.; et al. Transendocardial mesenchymal stem cells and mononuclear bone marrow cells for ischemic cardiomyopathy: The TAC-HFT randomized trial. JAMA 2014, 311, 62–73.
- 85.
Bartolucci, J.; Verdugo, F.J.; González, P.L.; et al. Safety and efficacy of the intravenous infusion of umbilical cord mesenchymal stem cells in patients with heart failure: A phase 1/2 randomized controlled trial (RIMECARD trial [randomized clinical trial of intravenous infusion umbilical cord mesenchymal stem cells on cardiopathy]). Circ. Res. 2017, 121, 1192–1204.
- 86.
Mathiasen, A.B.; Qayyum, A.A.; Jørgensen, E.; et al. Bone marrow-derived mesenchymal stromal cell treatment in patients with severe ischaemic heart failure: A randomized placebo-controlled trial (MSC-HF trial). Eur. Heart J. 2015, 36, 1744–1753.
- 87.
Perin, E.C.; Borow, K.M.; Henry, T.D.; et al. Randomized trial of targeted transendocardial mesenchymal precursor cell therapy in patients with heart failure. J. Am. Coll. Cardiol. 2023, 81, 849–863.
- 88.
Qayyum, A.A.; Frljak, S.; Juhl, M.; et al. Mesenchymal stromal cells to treat patients with non-ischaemic heart failure: Results from SCIENCE II pilot study. ESC Heart Fail. 2024, 11, 3882–3891.
- 89.
Perin, E.C.; Borow, K.M.; Henry, T.D.; et al. Mesenchymal precursor cells reduce mortality and major morbidity in ischaemic heart failure with inflammation: DREAM-HF. Eur. J. Heart Fail. 2025, 27, 3288–3296.
- 90.
Qayyum, A.A.; van Klarenbosch, B.; Frljak, S.; et al. Effect of allogeneic adipose tissue-derived mesenchymal stromal cell treatment in chronic ischaemic heart failure with reduced ejection fraction–the SCIENCE trial. Eur. J. Heart Fail. 2023, 25, 576–587.
- 91.
Chaaban, N.; Kastrup, J.; Qayyum, A.A. Results from the SCIENCE and Danish ASC trials using allogeneic mesenchymal stromal cells to treat ischemic heart failure patients. Regen. Med. 2025, 20, 573–584.
- 92.
Zhao, M.; Xue, Y.; Tian, Q.; et al. Impact of mesenchymal stem cell therapy on cardiac function and outcomes in acute myocardial infarction: A meta-analysis of clinical studies. Cell Transplant. 2025, 34, 09636897251359773.
- 93.
Kishino, Y.; Fukuda, K. Unlocking the pragmatic potential of regenerative therapies in heart failure with next-generation treatments. Biomedicines 2023, 11, 915.
- 94.
Xu, L.; Xu, M.; Sun, X.; et al. Quantitative comparison of gold nanoparticle delivery via the enhanced permeation and retention (EPR) effect and mesenchymal stem cell (MSC)-based targeting. Acs Nano 2023, 17, 2039–2052.
- 95.
Bahari, M.; Mokhtari, H.; Yeganeh, F. Stem cell therapy, the market, the opportunities and the threat. Int. J. Mol. Cell. Med. 2023, 12, 310.
- 96.
Sipp, D.; Robey, P.G.; Turner, L. Clear up this stem-cell mess. Nature 2018, 561, 455–457.
- 97.
Seyihoglu, B.; Orhan, I.; Okudur, N.; et al. 20 years of treating ischemic cardiomyopathy with mesenchymal stromal cells: A meta-analysis and systematic review. Cytotherapy 2024, 26, 1443–1457.
- 98.
Chowdhury, M.A.; Zhang, J.J.; Rizk, R.; et al. Stem cell therapy for heart failure in the clinics: New perspectives in the era of precision medicine and artificial intelligence. Front. Physiol. 2024, 14, 1344885.
- 99.
Johnston, P.V.; Raval, A.N.; Henry, T.D.; et al. Dare to dream? Cell-based therapies for heart failure after DREAM-HF: Review and roadmap for future clinical study. Am. Heart J. Plus Cardiol. Res. Pract. 2022, 13, 100118.
- 100.
Han, X.; Liao, R.; Li, X.; et al. Mesenchymal stem cells in treating human diseases: Molecular mechanisms and clinical studies. Signal Transduct. Target. Ther. 2025, 10, 262.
- 101.
Attar, A.; Bahmanzadegan Jahromi, F.; Kavousi, S.; et al. Mesenchymal stem cell transplantation after acute myocardial infarction: A meta-analysis of clinical trials. Stem Cell Res. Ther. 2021, 12, 600.
- 102.
Yu, J.; Zhang, R.; Mao, Y.; et al. Efficacy and safety of mesenchymal stem cell therapy in patients with acute myocardial infarction: A systematic review and meta-analysis of randomized controlled trials. Curr. Stem Cell Res. Ther. 2022, 17, 793–807.
- 103.
Wruck, W.; Graffmann, N.; Spitzhorn, L.-S.; et al. Human induced pluripotent stem cell-derived mesenchymal stem cells acquire rejuvenation and reduced heterogeneity. Front. Cell Dev. Biol. 2021, 9, 717772.
- 104.
Wu, Z.; Su, Y.; Li, J.; et al. Induced pluripotent stem cell-derived mesenchymal stem cells: Whether they can become new stars of cell therapy. Stem Cell Res. Ther. 2024, 15, 367.
- 105.
Thavapalachandran, S.; Le, T.Y.L.; Romanazzo, S.; et al. Pluripotent stem cell-derived mesenchymal stromal cells improve cardiac function and vascularity after myocardial infarction. Cytotherapy 2021, 23, 1074–1084.
- 106.
Chen, J.; Liang, X.; Han, Q.; et al. Exosomal miR-202-5p derived from iPSC-MSCs protects against myocardial infarction through inhibition of cardiomyocyte pyroptosis. Stem Cell Res. Ther. 2025, 16, 282.
- 107.
Bloor, A.J.; Patel, A.; Griffin, J.E.; et al. Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: A phase I, multicenter, open-label, dose-escalation study. Nat. Med. 2020, 26, 1720–1725.
- 108.
Watanabe, T.; La Shu, S.; del Rio-Espinola, A.; et al. Evaluating teratoma formation risk of pluripotent stem cell-derived cell therapy products: A consensus recommendation from the Health and Environmental Sciences Institute’s International Cell Therapy Committee. Cytotherapy 2025.
- 109.
Martin, R.M.; Fowler, J.L.; Cromer, M.K.; et al. Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 2020, 11, 2713.
- 110.
Kelly, K.; Bloor, A.J.; Griffin, J.E.; et al. Two-year safety outcomes of iPS cell-derived mesenchymal stromal cells in acute steroid-resistant graft-versus-host disease. Nat. Med. 2024, 30, 1556–1558.
- 111.
Kang, K.; Ma, R.; Cai, W.; et al. Exosomes secreted from CXCR4 overexpressing mesenchymal stem cells promote cardioprotection via Akt signaling pathway following myocardial infarction. Stem Cells Int. 2015, 2015, 659890.
- 112.
Huang, Y.; Li, X.; Yang, L. Hydrogel encapsulation: Taking the therapy of mesenchymal stem cells and their derived secretome to the next level. Front. Bioeng. Biotechnol. 2022, 10, 859927.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com