Background: Caloric restriction (CR) is a powerful non-pharmacologic intervention known to extend lifespan and improve cardiovascular health. Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) exert antihypertrophic and antifibrotic effects through natriuretic peptide receptor 1 (NPR1), a guanylyl cyclase receptor that generates cyclic GMP (cGMP). Whether intact NPR1 signaling is required for the cardioprotective effects of CR remains unknown. Methods: NPR1 knockout (KO), heterozygous KO (+/−), and wild-type (WT) littermate mice were subjected to a CR regimen (20% caloric reduction for 2 weeks, then 40% for 2 weeks). WT mice were subjected to transverse aortic constriction (TAC) to model acute pressure overload, whereas NPR1-deficient mice were studied in the setting of established genetic cardiomyopathy. Cardiac structure, function, fibrosis, apoptosis, ATP levels, oxidative and ER stress, and signaling pathways (cGMP, eNOS/NO/sGC) were assessed by echocardiography, histology, biochemical assays, and Western blotting. Results: In WT mice, CR significantly attenuated TAC-induced hypertrophy, preserved cardiac function, reduced fibrosis, and decreased oxidative and ER stress. In contrast, CR did not attenuate or reverse established NPR1-deficiency-induced hypertrophy, fibrosis, or dysfunction, despite reducing oxidative stress and ER stress. CR preserved ATP content in NPR1 KO hearts, but cGMP levels remained profoundly depressed (~90% reduction). Compensatory activation of NO-sGC signaling was observed in NPR1 KO hearts, but this response was insufficient to restore myocardial cGMP levels or limit structural remodeling. Conclusions: These findings indicate that intact NPR1-cGMP signaling is required for CR-mediated protection against pressure-overload–induced cardiac remodeling, whereas CR alone is insufficient to reverse established, genetically programmed cardiomyopathy in the absence of NPR1. Although CR reduces oxidative stress, ER stress, and preserves myocardial ATP, these adaptations are insufficient to compensate for the loss of NPR1-cGMP signaling in reversing established pathological cardiac remodeling. Thus, the cardioprotective efficacy of CR appears to be context-dependent and may require intact natriuretic peptide signaling.
- Open Access
- Article
Caloric Restriction Attenuates Oxidative Injury but Fails to Reverse Established Cardiac Remodeling in Atrial Natriuretic Peptide Receptor 1-Deficient Mice †
- Kai Chen 1,‡,
- Derek Timm 1,
- Yuan Huang 1,2,
- Satoru Kobayashi 1,2,
- Qiangrong Liang 1,2,*
Author Information
Received: 23 Dec 2025 | Revised: 02 Feb 2026 | Accepted: 02 Feb 2026 | Published: 29 Jun 2026
Abstract
Keywords
caloric restriction (CR) | natriuretic peptide receptor 1 (NPR1) | cardiac remodeling | cGMP signaling | oxidative stress | pressure overload (TAC)
1. Introduction
Caloric restriction (CR), defined as a chronic reduction in caloric intake without malnutrition, is one of the most reproducible interventions known to extend lifespan and delay age-related disease across species, from yeast to primates [1,2,3,4,5,6,7,8,9,10,11,12,13]. In humans, CR improves cardiovascular risk profiles, delays cardiovascular aging, and enhances cardiac stress resistance in both healthy and at-risk populations [9,14,15,16]. Specifically, CR lowers blood pressure, improves insulin sensitivity, favorably alters lipid profiles, and suppresses atherosclerosis [7,8,17,18,19,20]. CR also reduces cardiac mass and ventricular stiffness, thereby improving diastolic function in humans [15,21,22]. Consistent with these findings, animal studies show that CR attenuates age-associated left ventricular hypertrophy, limits oxidative injury, preserves diastolic function [23,24,25,26], and protects against acute ischemic injury and chronic maladaptive cardiac remodeling [27,28,29,30,31]. Together, evidence from both human and animal studies underscores CR as a powerful intervention for preserving cardiac structure and function and mitigating age- and stress-induced heart disease.
The mechanistic underpinnings of CR’s cardioprotective effects are multifaceted. At the cellular level, CR enhances mitochondrial efficiency, increases autophagy, and reduces reactive oxygen species production. CR also activates nutrient-sensing pathways such as AMP-activated protein kinase (AMPK) [32,33] and sirtuins (SIRT1, SIRT3) [34], and suppresses mTOR signaling [35], thereby promoting cellular stress resistance. These adaptations collectively maintain energetic homeostasis and protect against maladaptive remodeling. However, the interplay between CR and cardiac hormone systems, particularly natriuretic peptides, remains incompletely understood.
Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are cardiac hormones released primarily in response to atrial and ventricular stretch, respectively. Initially identified for their natriuretic and vasodilatory functions, both peptides are now recognized as key regulators of cardiac remodeling [33,36]. Their effects are mediated through natriuretic peptide receptor 1 (NPR1), also known as guanylyl cyclase-A, GC-A, a receptor guanylyl cyclase that generates cGMP upon ligand binding [36]. Activation of the downstream cGMP signaling pathway engages protein kinase G (PKG), cyclic nucleotide-gated channels, and phosphodiesterases, resulting in vasodilation, natriuresis, inhibition of renin-angiotensin-aldosterone signaling, and direct antihypertrophic actions in cardiomyocytes [37]. Genetic ablation of NPR1 in mice leads to salt-sensitive hypertension, marked cardiac hypertrophy, interstitial fibrosis, and early mortality [38,39,40], underscoring its essential role in cardiovascular homeostasis. Mechanistically, NPR1 signaling counteracts prohypertrophic transcriptional programs including NFAT, NF-κB, and AP-1 [41], and suppresses profibrotic mediators such as TGF-β and collagen synthesis [39]. In addition, NPR1 activation also reduces oxidative stress by improving mitochondrial function and limiting NADPH oxidase activity [42,43]. Collectively, ANP/BNP-NPR1 signaling constitutes a fundamental protective axis that restrains pathological cardiac remodeling.
Evidence indicates that caloric restriction (CR) interacts with the natriuretic peptide system. Fasting or short-term CR elevates circulating ANP concentrations and augments renal and hemodynamic sensitivity to exogenously administered ANP in humans [44,45]. These adaptations may contribute to CR’s capacity to mitigate pathological cardiac remodeling during pressure overload or metabolic stress. However, whether NPR1 signaling is required for the cardioprotective effects of CR has not been directly examined. Given that CR activates multiple, partially redundant stress-resistance pathways, including AMPK, sirtuins, and NO-cGMP signaling, it remains possible that CR preserves cardioprotection even in the absence of NPR1. Conversely, NPR1 signaling may be indispensable, with CR’s beneficial effects converging on ANP/BNP-NPR1 signaling as a central integrative hub.
In this study, we tested the hypothesis that intact NPR1 is required for CR-mediated protection against pathological cardiac remodeling, particularly in the setting of acute hemodynamic stress. Using NPR1 knockout, heterozygous, and wild-type mice, we evaluated whether a four-week CR regimen confers protection against cardiac hypertrophy, fibrosis, and dysfunction. We also assessed myocardial energetic status (ATP content), oxidative and endoplasmic reticulum stress, and compensatory NO-sGC signaling to distinguish NPR1-dependent from NPR1-independent mechanisms underlying the cardioprotective effects of CR.
2. Materials and Methods
2.1. Experimental Animals
Natriuretic peptide receptor 1 (NPR1) gene-targeted mice were obtained from the Jackson Laboratory (Bar Harbor, ME 04609, USA, Stock no. 004374). The mutant line was originally generated on a 129 background and subsequently backcrossed onto the C57BL/6 background for more than eight generations. Homozygous NPR1 knockout (KO), heterozygous (NPR1+/−), and wild-type (WT) littermates were produced by in-house breeding of a colony maintained in our institutional animal facility. Both male and female mice aged 12 weeks were used, with sex balanced across experimental groups. All procedures were approved by the Institutional Animal Care and Use Committee and conducted in accordance with NIH guidelines.
2.2. Caloric Restriction Protocol
Caloric restriction (CR) was implemented as described [33]. Twelve-week-old mice were fed the #2018 rodent diet ad libitum (AL) for one week to establish baseline food intake. Average daily consumption during this period was used to calculate caloric allocation for the caloric restriction (CR) group. Mice were then randomly assigned to AL or CR groups. AL mice continued to receive food ad libitum, whereas CR mice were fed 20% less than AL intake for two weeks, followed by 40% less for an additional two weeks. CR mice received their daily food allotment at 5:00 p.m. Food allocation was adjusted weekly based on consumption in the AL group. When indicated, transverse aortic constriction (TAC) was performed during the second week of the caloric restriction (CR) protocol. At study completion, cardiac function was assessed by echocardiography. Mice were subsequently euthanized, blood was collected, and hearts were rapidly excised, rinsed in cold saline, weighed, and sectioned for downstream analyses.
2.3. Transverse Aortic Constriction (TAC)
To induce pressure overload, mice underwent transverse aortic constriction (TAC) surgery under inhalational anesthesia (1.5–2.5% isoflurane in 1 L/min oxygen). Body temperature was continuously monitored using a rectal thermometer and maintained with a heating platform. A 27-gauge needle was placed adjacent to the transverse aorta between the innominate and left common carotid arteries, and a 7-0 silk suture was tied securely around both the needle and the aorta. The needle was then removed to create a standardized constriction. Sham-operated mice underwent the same surgical procedure without aortic ligation. Postoperative analgesia was provided by intraperitoneal administration of buprenorphine (0.1 µg/g body weight).
2.4. Echocardiography
Transthoracic echocardiography was performed at baseline and 4 weeks after TAC under anesthesia (3% isoflurane induction, 1.5% maintenance) using a high-resolution ultrasound system (Vevo 2100, VisualSonics, Toronto, ON M4N3N1, Canada) as described previously [33]. Left ventricular (LV) end-diastolic diameter (LVEDD), end-systolic diameter (LVESD), fractional shortening (FS), and ejection fraction (EF) were measured from 2-D short-axis M-mode tracings at the level of the papillary muscle. All analyses were performed by investigators blinded to genotype and treatment.
2.5. Histology and Morphometry
Immediately after harvest, a mid-ventricular myocardial slice was fixed in 4% (w/v) paraformaldehyde overnight, embedded in paraffin, and sectioned at 5 µm by the Histology Core at the Burnham Institute for Medical Research (La Jolla, CA, USA). Fibrosis was assessed by Masson’s trichrome staining. High-resolution images were acquired using the Aperio Scanscope system (Burnham Institute, La Jolla, CA, USA), and cardiac fibrosis was quantified using ImageJ 2.0.0-beta-7 (NIH). For cardiomyocyte cross-sectional area analysis, a separate mid-ventricular myocardial slice was embedded in OCT compound (Sakura Finetek, Inc. Torrance, CA 90501, USA). Frozen ventricular sections (5 µm) were stained with wheat germ agglutinin (WGA; Sigma-Aldrich, St. Louis, MO, USA) to delineate cardiomyocyte membranes and Alexa Fluor 488–conjugated isolectin IB4 (Thermo Fisher Scientific, Waltham, MA, USA) to label capillaries. Images were captured at 400× magnification using an FV1000 confocal microscope (Olympus, Tokyo 192-8507, Japan). Cardiomyocyte cross-sectional areas were quantified using ImageJ from 4–5 regions per heart, encompassing at least 1000 cardiomyocytes per heart.
2.6. ATP Measurement
Myocardial ATP content was measured using a luciferase-based ATP determination kit (Thermo Fisher). Cardiac tissue samples were homogenized in ice-cold assay buffer according to the manufacturer’s instructions, and ATP levels were quantified by luminescence. Values were normalized to total protein concentration.
2.7. cGMP Assay
Tissue cyclic GMP (cGMP) levels were measured using a cGMP complete ELISA kit (Enzo Life Sciences, Farmingdale, NY, USA). Hearts were rapidly excised, snap‑frozen in liquid nitrogen, and pulverized to preserve endogenous cGMP. The powdered tissue was homogenized in 0.1 M HCl to inactivate phosphodiesterases and prevent cGMP degradation. After centrifugation, supernatants were collected and processed according to the manufacturer’s instructions. The absorbances were read at 405 nm using a SynergyTM Mx multi-mode microplate reader from BioTek (Winooski, VT, USA). The concentrations were calculated based on a standard curve generated at the same time.
2.8. Nitric Oxide (NO) Assay
Nitric oxide (NO) levels were measured using the Abcam Nitric Oxide Assay Kit (ab272517). Because NO rapidly oxidizes to nitrite (NO₂⁻) and nitrate (NO3⁻), total nitrite/nitrate was quantified as an index of NO production. Nitrate was enzymatically reduced to nitrite, followed by colorimetric detection using the Griess reaction. Absorbance was read at 540 nm, and NO levels were calculated from a sodium nitrite standard curve.
2.9. sGC Assay
Soluble guanylate cyclase (sGC) levels were quantified using a mouse soluble guanylate cyclase subunit α‑3 ELISA kit (MyBioSource, San Diego, CA, USA). Tissue samples were processed according to the manufacturer’s instructions, and sGC concentrations were calculated from a standard curve generated in parallel.
2.10. Western Blot Analyses
Protein expression was assessed by immunoblotting using standard procedures. Briefly, heart tissues were homogenized in ice-cold RIPA buffer supplemented with protease and phosphatase inhibitor cocktails (Roche, Basel, Switzerland). Lysates were clarified by centrifugation at 12,000× g for 15 min at 4 °C, and protein concentrations were determined using a BCA assay (Thermo Fisher Scientific). Equal amounts of protein (20–40 µg) were resolved on SDS–polyacrylamide gels and transferred onto PVDF membranes (Millipore, Burlington, MA, USA). Membranes were blocked for 1 h at room temperature in Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% non-fat dry milk, as indicated. Membranes were then incubated overnight at 4 °C with primary antibodies diluted in blocking buffer.
The following primary antibodies were used: CHOP, BiP, protein disulfide isomerase (PDI), PERK, IRE1α, and GAPDH (all from Cell Signaling Technology, Danvers, MA, USA; typically 1:1000 for target proteins and 1:50,000 for GAPDH); Atp5g2, cytochrome c oxidase subunit IV (Cox IV), cytochrome b5 (Cytb5), optic atrophy 1 (Opa1), atrial natriuretic peptide (ANP), and natriuretic peptide receptor-A (NPR-A/NPR1) (Abcam, Cambridge, UK; typically 1:1000); dynamin-related protein 1 (Drp1) (Santa Cruz Biotechnology, Dallas, TX, USA; 1:500–1:1000); and oxidative stress markers 4-hydroxynonenal (4-HNE) and nitrotyrosine (Millipore; 1:1000). CHOP (~29 kDa), BiP (~75 kDa), PDI (~57 kDa), PERK (~120–140 kDa), IRE1α (~110–130 kDa), eNOS and phospho-eNOS (Ser1177; ~135 kDa), Drp1 (~80 kDa), Cox IV (~17 kDa), Cytb5 (~15 kDa), and Atp5g2 (~9 kDa) were detected at their expected molecular weights. Opa1 appeared as multiple bands (~90–110 kDa) corresponding to long and short isoforms. Oxidized proteins, 4-HNE and nitrotyrosine immunoblots showed multiple bands reflecting protein adduct formation.
After primary antibody incubation, membranes were washed three times with TBST and incubated for 1 h at room temperature with appropriate horseradish peroxidase–conjugated secondary antibodies (anti-rabbit or anti-mouse IgG; Jackson ImmunoResearch, West Grove, PA, USA) diluted 1:5000 in blocking buffer. Immunoreactive bands were visualized using enhanced chemiluminescence (ECL; Thermo Fisher Scientific) and imaged with a ChemiDoc imaging system 3000 (Bio-Rad, Hercules, CA, USA).
Band intensities were quantified using ImageJ software (NIH). Target protein expression was normalized to GAPDH whose expression is not changed across genotypes and dietary conditions in this study.
2.11. Oxidative Stress and ER Stress Assays
Protein carbonyl content in the heart homogenates was measured by Western blot analysis using the Oxyblot™ Protein Oxidation Detection Kit (S7150, Chemicon, Temecula, CA, USA), based on immunochemical detection of protein carbonyl groups derived from 2,4-dinitrophenyl hydrazine [33]. Lipid peroxidation and protein nitration were evaluated by standard Western blotting procedure using 4-Hydroxynonenal (4-HNE) and 3-Nitrotyrosine (3-NT) antibodies respectively. ER stress markers (CHOP, GRP78 or BiP, PDI, PERK and IRE1α) were measured by Western blot analyses.
2.12. RNA isolation, RT, and Quantitative PCR Analyses
Total RNA was isolated using the TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the recommendations of the manufacturer. A total of 1 μg RNA from each sample was used for RT using TaqMan® Reverse Transcription Reagents (Invitrogen). The cDNA was diluted by 10 using ddH2O and used for quantitative PCR analysis (Invitrogen). The primer sequences are as follows. ANP forward: ATGGGCTCCTTCTCCATCA, and reverse GGAAGCTGTTGCAGCCTAGT; BNP forward: AAGGTGCTGTCCCAGATGATT, and reverse: TTCAGTGCGTTACAGCCCAAA; GAPDH forward: AAGGTCATCCCAGAGCTGAAC, and reverse: TCATTGAGAGCAATGCCAGCC. Gene expression was quantified by GelDoc XR+ imaging system (Bio-Rad).
2.13. Statistical Analysis
Data are presented as mean ± SEM. Comparisons among multiple groups were performed using two-way ANOVA with genotype and diet as factors, followed by Bonferroni post hoc testing to correct for multiple comparisons, as implemented in GraphPad Prism 6. A two-tailed p value < 0.05 was considered statistically significant. Some analyses may not have been powered to detect small effect sizes, including potential haploinsufficiency; therefore, negative findings should be interpreted with appropriate caution.
3. Results
3.1. CR is Sufficient to Attenuate Pressure Overload-Induced Cardiac Remodeling and Functional Impairment in Wild Type C57/6 Mice
To confirm the ability of caloric restriction (CR) to modulate the cardiac remodeling response, wild-type C57BL/6 mice were subjected to CR for 4 weeks (20% and 40% food reduction, each applied for 2 weeks), with transverse aortic constriction (TAC) performed during week 2. Following TAC, mice fed ad libitum (AL) developed robust left ventricular (LV) hypertrophy, as evidenced by a 42% increase in the heart weight-to-body weight (HW/BW) ratio compared with sham controls (Figure 1A, p < 0.01). CR significantly blunted the TAC-induced hypertrophic response, limiting the increase in HW/BW to 18% (Figure 1A, p < 0.05 vs. AL).
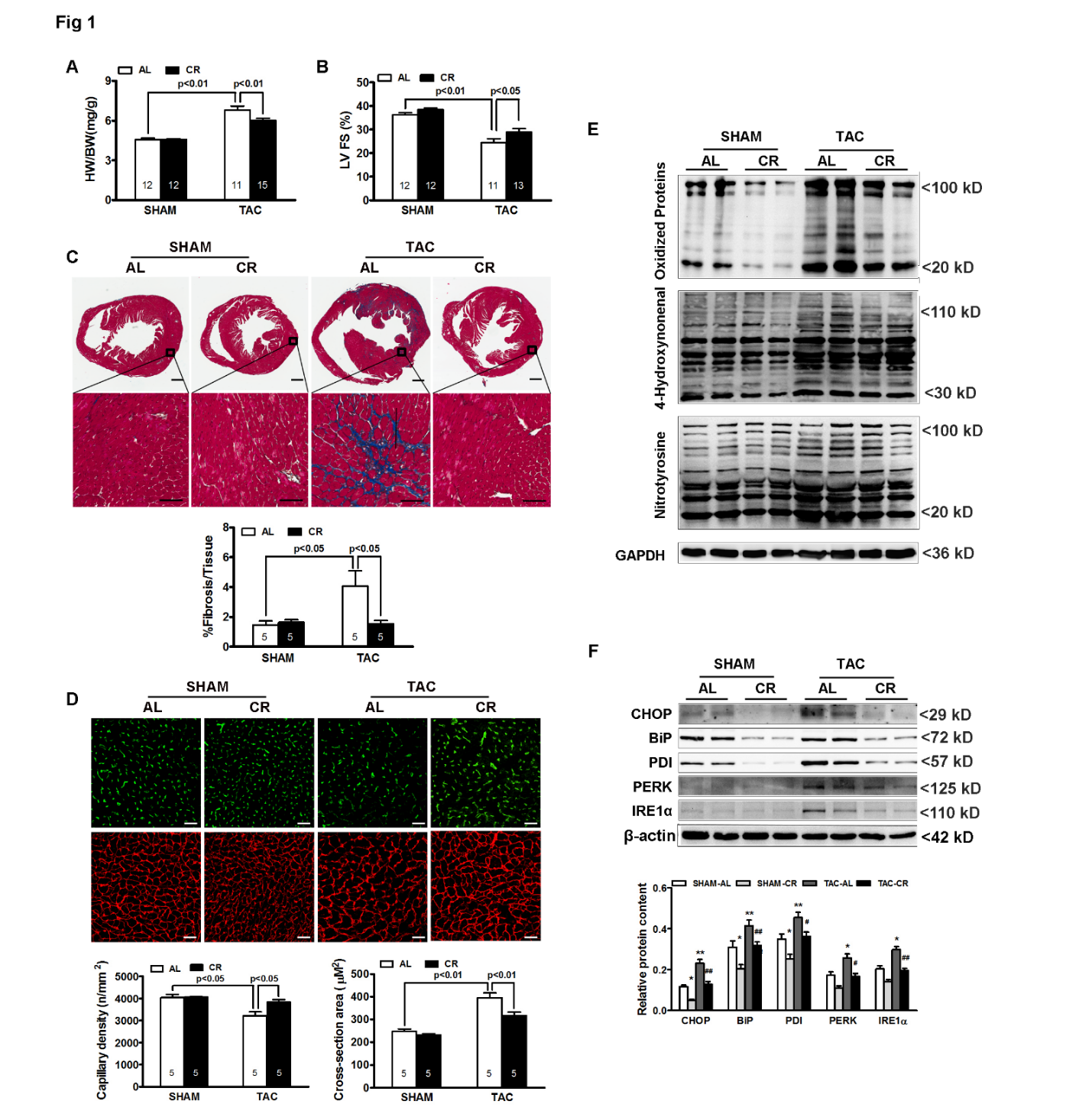
Echocardiographic analysis further demonstrated that TAC significantly reduced LV fractional shortening (FS) in AL mice, whereas CR preserved systolic function (Figure 1B; 33 ± 2% vs. 26 ± 3%, p < 0.05). Consistent with these functional changes, Masson’s trichrome staining revealed marked interstitial and perivascular fibrosis in TAC hearts from AL mice (Figure 1C, fibrotic area: 12 ± 1% vs. 4 ± 1% in sham, p < 0.01), which was significantly attenuated by CR (6 ± 1%, p < 0.05 vs. AL). In addition, TAC increased cardiomyocyte cross-sectional area and reduced capillary density, both of which were partially reversed by CR (Figure 1D).
At the molecular level, Western blot analyses showed that TAC markedly increased oxidative stress, as indicated by elevated levels of oxidized protein carbonyls, 4-hydroxynonenal, and nitrotyrosine (Figure 1E). CR significantly reduced these oxidative stress markers in TAC hearts. Markers of endoplasmic reticulum (ER) stress, including CHOP, BiP, PDI, PERK, and IRE1α, were also markedly induced by TAC (Figure 1F). CR blunted the induction of these ER stress markers.
Collectively, these results demonstrate that CR is sufficient to attenuate pressure overload-induced cardiac remodeling and functional impairment in mice, and that these protective effects are associated with reduced oxidative stress and ER stress.
3.2. CR Does Not Attenuate Established Cardiac Remodeling in NPR1 Deficient Mice
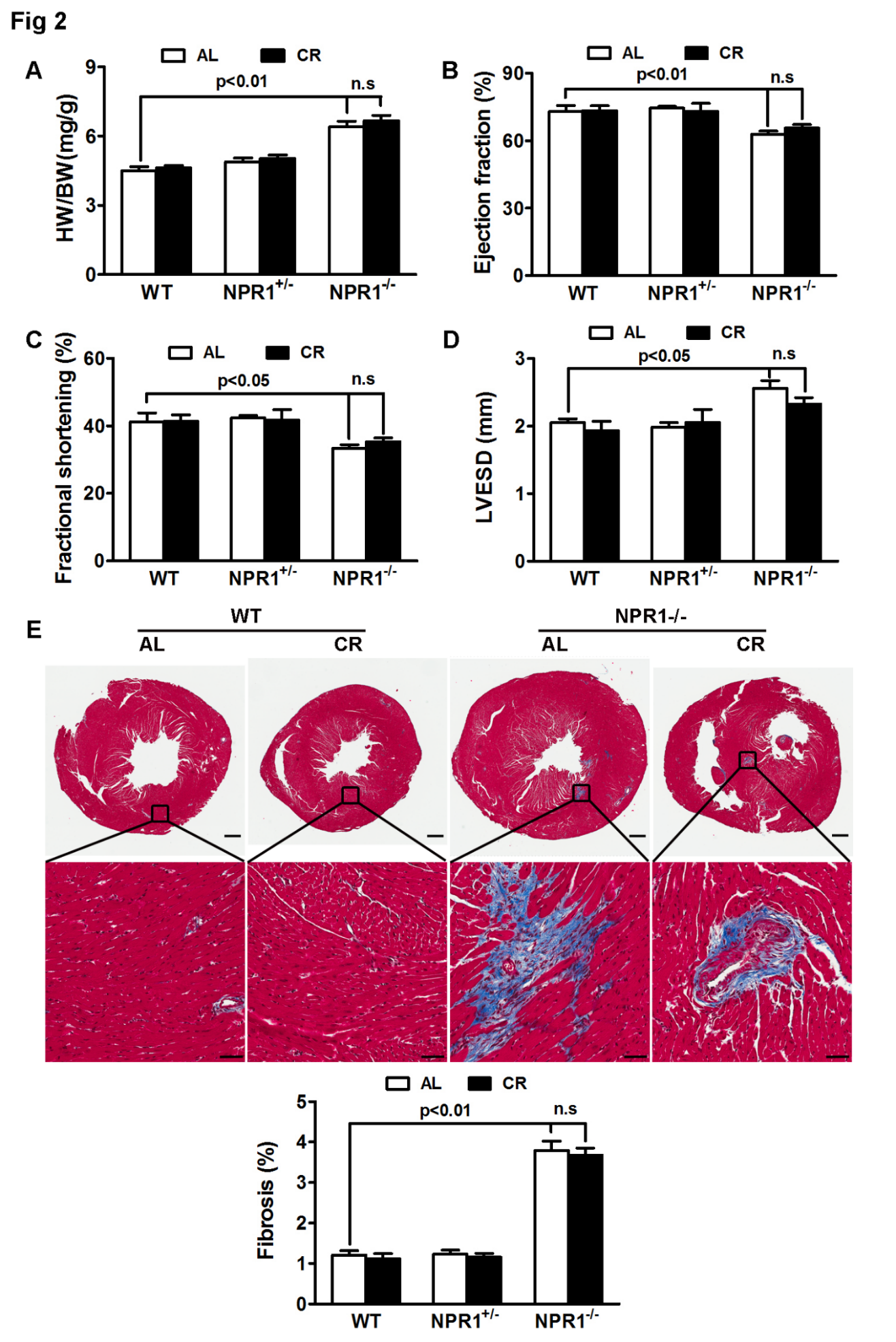
Genetic ablation of NPR1 in mice leads to cardiac hypertrophy and interstitial fibrosis [38,39,40]. Given the ability of CR to attenuate TAC-induced cardiac remodeling (Figure 1), we hypothesized that CR might also mitigate cardiac remodeling in NPR1 deficient mice. As shown in Figure 2, no significant difference was observed between WT and NPR1+/− mice in HW/BW ratio (Figure 2A), fractional shortening (FS, Figure 2C), ejection fraction (EF, Figure 2B), left ventricular end-systolic diameter (LVESD, Figure 2D), or fibrosis (Figure 2E). In contrast, NPR1 KO mice exhibited marked cardiac hypertrophy and fibrosis accompanied by impaired cardiac function compared with WT mice.
However, in contrast to its effects on TAC-induced cardiac hypertrophy, CR neither attenuated nor reversed established cardiac remodeling in NPR1 KO mice, as none of the measured parameters differed significantly between ad libitum (AL)- and CR-fed KO animals. Collectively, these findings indicate that, unlike its protective effects against TAC-induced cardiac remodeling, CR is ineffective in mitigating cardiac remodeling driven by NPR1 deficiency. Because NPR1 KO mice develop a congenital, developmentally programmed cardiomyopathy prior to the initiation of CR, these results reflect an inability of CR to reverse established disease rather than a failure to prevent disease onset.
3.3. CR Reduces Oxidative Stress and ER Stress Independent of NPR1
Western blot analyses revealed markedly increased oxidative stress in NPR1 KO hearts, as indicated by elevated levels of oxidized protein carbonyls, 4-hydroxynonenal, and nitrotyrosine, reflecting oxidative damage (Figure 3A). Notably, CR significantly reduced these oxidative stress markers in WT, NPR1+/−, and NPR1 KO mice alike.
ER stress markers, including CHOP, BiP, PDI, PERK, and IRE1α, were also markedly elevated in NPR1 KO hearts (Figure 3B). CR blunted the induction of these ER stress markers across all genotypes, indicating that the stress-reducing effects of CR occur independently of NPR1.
Collectively, these findings demonstrate that although CR robustly suppresses oxidative and ER stress regardless of NPR1 status, these adaptive responses alone are insufficient to prevent cardiac remodeling in NPR1-deficient mice.
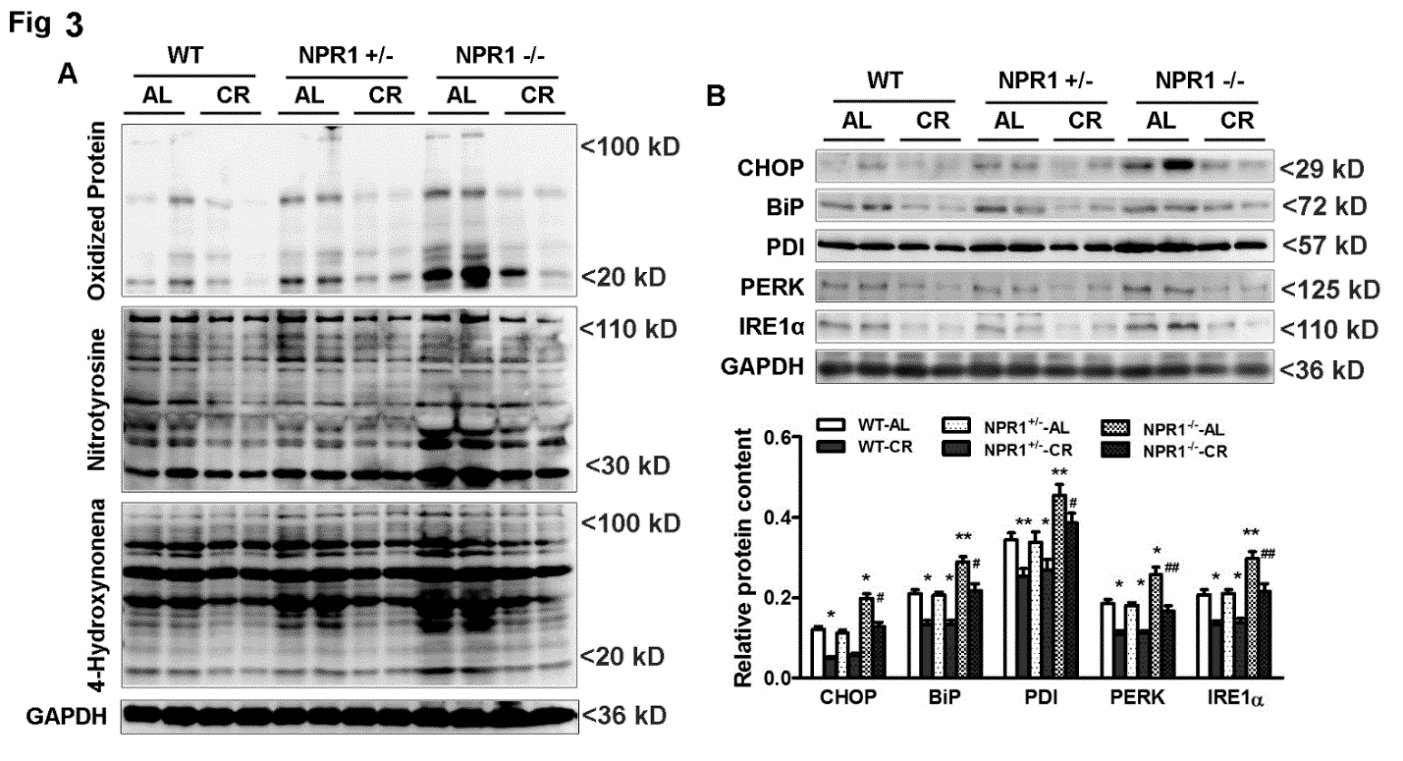
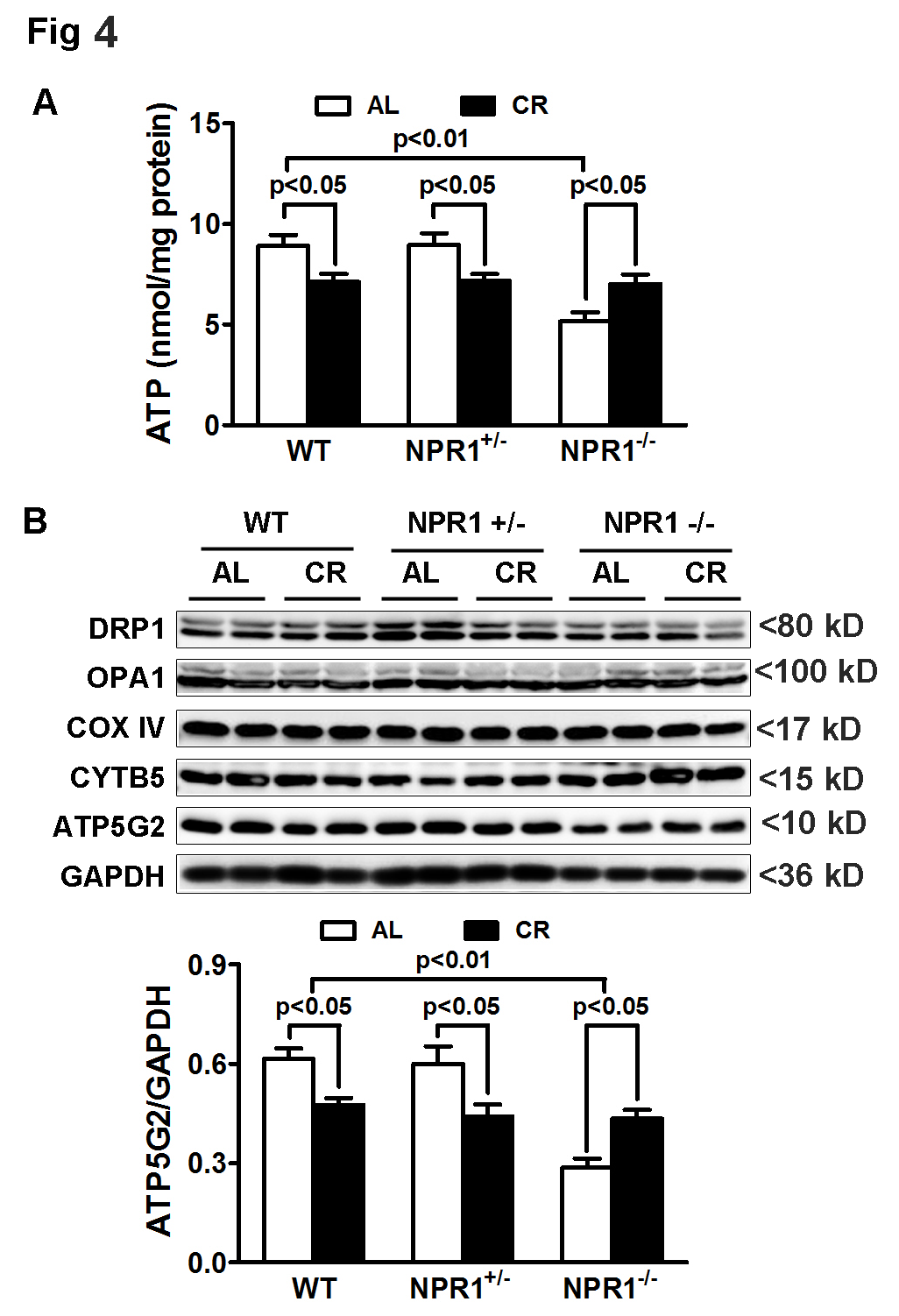
3.4. CR Preserves Myocardial ATP Content Independent of NPR1
ATP content was significantly reduced in NPR1 KO hearts compared with WT and NPR1+/− hearts. Interestingly, CR reduced ATP levels in WT and NPR1+/− hearts but did not further decrease ATP content in NPR1 KO hearts. Instead, ATP levels in NPR1 KO mice were higher under ad libitum (AL) feeding than under CR (Figure 4A), a pattern associated with preserved expression of the mitochondrial ATP synthase subunit ATP5G2 (Figure 4B). These findings suggest that, despite persistent hypertrophy and cardiac dysfunction, myocardial ATP levels are maintained in NPR1-deficient mice under CR. Together, these results indicate that CR preserves myocardial energetics through NPR1-independent mechanisms, potentially via enhanced mitochondrial efficiency and reduced metabolic demand.
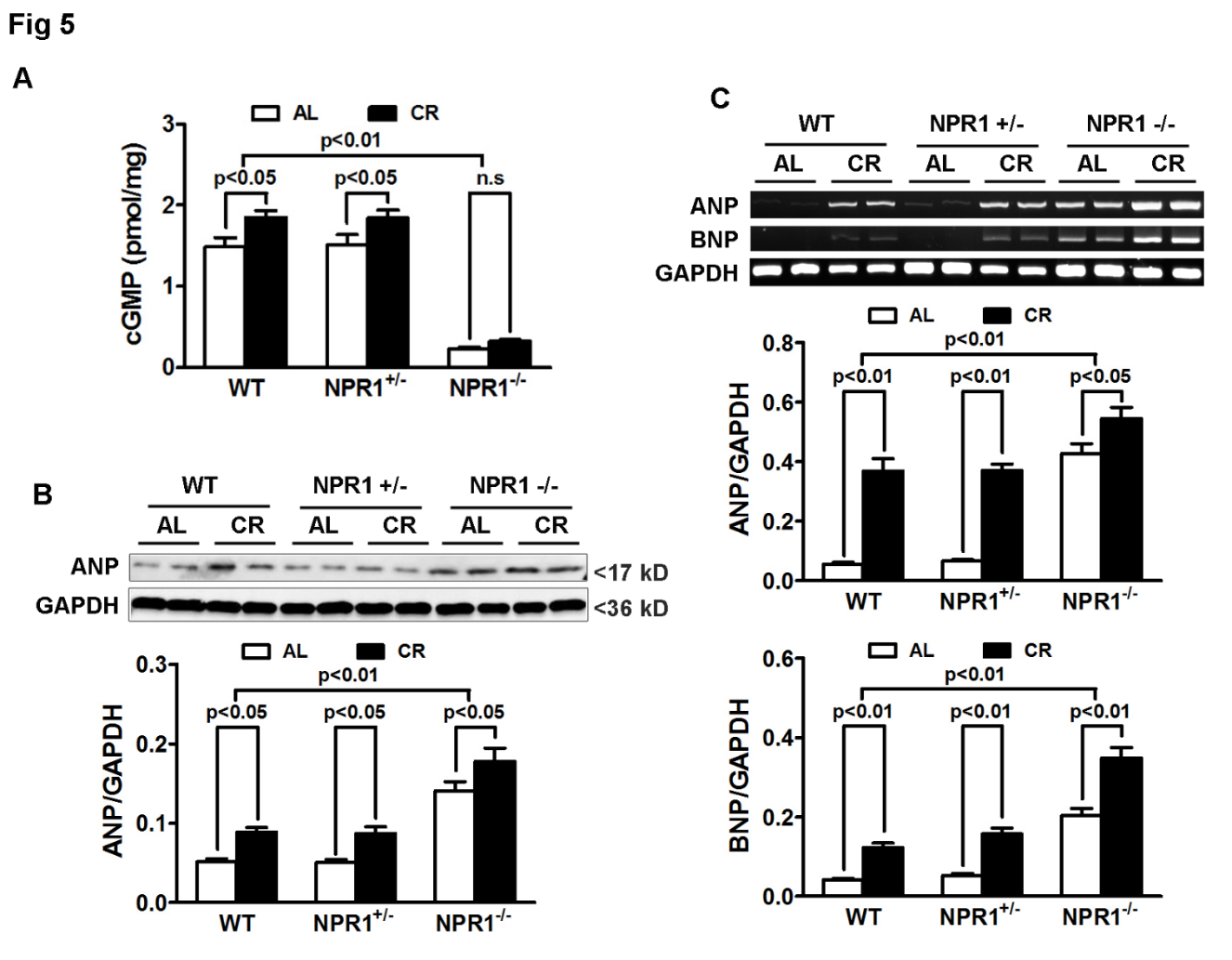
3.5. cGMP Signaling Is Severely Impaired in NPR1 KO Mice Despite CR
To directly assess downstream signaling, myocardial cGMP levels were measured across genotypes and dietary conditions. CR increased cGMP content by approximately 20% in both WT and NPR1+/− hearts compared with ad libitum (AL) feeding. In contrast, NPR1 knockout (KO) mice exhibited profoundly reduced basal cGMP levels (~90% reduction compared with WT), which were not restored by CR (Figure 5A). This persistent loss of cGMP signaling in NPR1 KO hearts correlated with the absence of CR-mediated antihypertrophic and antifibrotic effects, supporting a context-dependent requirement for intact NPR1-cGMP signaling in mediating CR-associated suppression of pathological remodeling.
3.6. ANP and BNP Are Upregulated in NPR1 KO Mice
Western blot analyses revealed an approximately 2.5-fold increase in atrial natriuretic peptide (ANP) protein levels in NPR1 KO mice compared with WT controls (p < 0.01). CR increased ANP protein expression by ~40% in both WT and NPR1+/− hearts and further elevated ANP levels in NPR1 KO hearts, likely reflecting a compensatory feedback upregulation of natriuretic peptide synthesis in response to impaired downstream signaling (Figure 5B).
Consistent with these findings, reverse transcription-polymerase chain reaction (RT-PCR) analyses demonstrated significantly increased ANP mRNA expression in NPR1 KO hearts, which was further augmented by CR. Similarly, BNP mRNA levels were markedly elevated in NPR1 KO hearts and were further induced by CR (Figure 5C). Together, these results suggest that natriuretic peptide expression is strongly upregulated in response to NPR1 deficiency and CR; however, in the absence of functional NPR1-cGMP signaling, this compensatory increase in ANP and BNP is insufficient to confer cardioprotection.
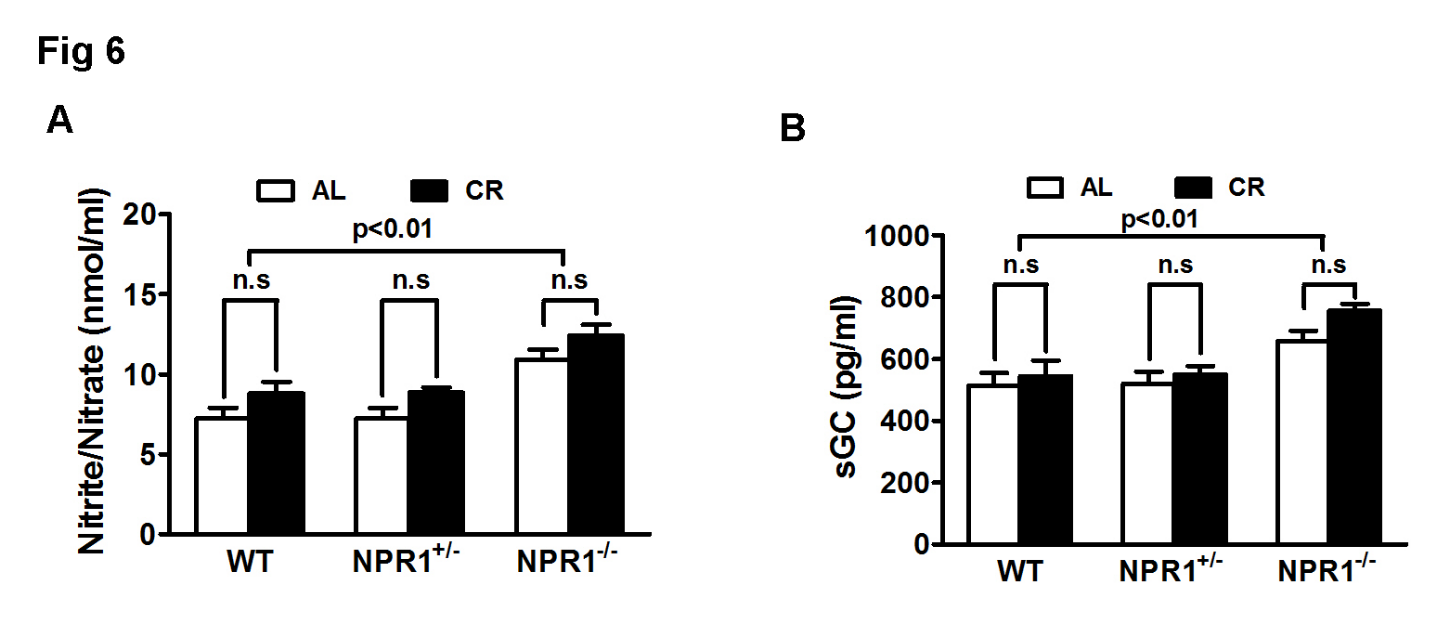
3.7. Compensatory Activation of NO-sGC Signaling in NPR1 KO Mice
The total amounts of nitrite (NO2−) and nitrate (NO3−) were significantly increased in NPR1 KO hearts (Figure 6A), suggesting an enhanced production of nitric oxide (NO). In parallel, expression of soluble guanylyl cyclase (sGC) was upregulated (Figure 6B), suggesting a compensatory attempt to restore cGMP signaling through the NO-sGC pathway. Despite these adaptations, myocardial cGMP levels remained profoundly reduced. Thus, although CR engages compensatory NO-sGC signaling in NPR1 KO mice, this response is insufficient to restore cGMP signaling or prevent pathological cardiac remodeling.
4. Discussion
The present study shows the ability of caloric restriction (CR) to attenuate pressure-overload-induced cardiac remodeling, consistent with a previous study in the spontaneously hypertensive rats [26]. In contrast, our results also demonstrate that CR fails to attenuate or reverse established cardiac hypertrophy, fibrosis, and functional decline in NPR1-deficient mice, despite preservation of myocardial ATP content and reduction of oxidative stress and ER stress. These findings indicate that NPR1-cGMP signaling represents a critical pathway for CR-mediated suppression of cardiac remodeling in the setting of acute pressure overload, while also highlighting the limited capacity of CR to reverse advanced, genetically programmed cardiomyopathy.
4.1. NPR1 as a Central Node in CR-Mediated Cardioprotection
CR exerts broad systemic and cellular effects, including improved mitochondrial efficiency, enhanced autophagy, reduced ROS generation, and suppression of nutrient-sensitive pathways such as mTOR. These adaptations are thought to provide redundant layers of protection against pathological remodeling. Our findings, however, reveal that the antihypertrophic and antifibrotic effects of CR are critically dependent on NPR1 signaling, even when oxidative stress, ER stress, and ATP content are favorably regulated. The requirement for NPR1 likely reflects its unique role in directly modulating cardiomyocyte growth and fibroblast activity. NPR1-cGMP-PKG signaling inhibits calcineurin-NFAT, MAPK, and hypertrophic transcriptional programs, while simultaneously suppressing profibrotic TGF-β/Smad signaling. These pathways converge on gene expression programs that govern hypertrophy and extracellular matrix deposition, outcomes that cannot be fully mitigated by improved mitochondrial energetics or reduced oxidative burden alone. This hierarchy suggests that CR’s activation of energy-sensing pathways (e.g., AMPK, sirtuins) and stress resistance mechanisms provides a supportive environment, but NPR1 signaling is required to directly suppress the maladaptive transcriptional responses to mechanical overload.
The natriuretic peptide receptor (NPR) family comprises NPR1 (GC-A), which primarily mediates ANP/BNP-induced cGMP production in the heart, NPR2 (GC-B), which preferentially responds to CNP, and NPR3, which functions largely as a clearance receptor. In this context, the potential roles of NPR2 and NPR3 in caloric restriction and NPR1 deficiency warrant brief discussion. Although compensatory changes in NPR2 or NPR3 could theoretically influence natriuretic peptide availability, the ~90% reduction in myocardial cGMP observed in NPR1 KO hearts strongly supports NPR1 as the dominant source of cardiomyocyte cGMP under these conditions. Accordingly, the observed phenotype is most consistent with loss of NPR1 signaling rather than redistribution within the natriuretic peptide receptor family, which remains an important area for future investigation.
Although heterozygous NPR1 KO (Npr1+/−) mice have been reported to exhibit impaired renal and metabolic responses under stress [46], we did not observe a significant cardiac phenotype in Npr1+/− mice compared with wild-type controls. Across all measured parameters, including NPR1 protein abundance, myocardial cGMP levels, and ANP protein levels (Figure 5), Npr1+/− mice were indistinguishable from wild-type mice, indicating preserved NPR1 signaling and no evidence of cardiac haploinsufficiency under baseline and CR conditions. Because these studies were performed in young adult mice (12 weeks old), age- or stress-dependent haploinsufficiency in older animals cannot be excluded.
4.2. Dissociation of Oxidative/ER Stress from Cardiac Remodeling
A critical observation in our data is the dissociation between "stress markers" and "structural remodeling". CR robustly reduced protein carbonyls, 4-hydroxynonenal (4-HNE), and nitrotyrosine levels, and blunted the induction of ER stress markers (CHOP, BiP, PERK) in all genotypes, including the NPR1 KO. While oxidative and ER stress are often cited as primary drivers of cardiac pathology, our results indicate that suppressing these factors is insufficient to prevent remodeling if cGMP signaling is absent. This suggests that while CR creates a more favorable intracellular environment, the “stop signal” for hypertrophic gene programs and fibroblast activation requires the NPR1-cGMP pathway. In the absence of NPR1, the heart remains committed to a remodeling program despite a significant reduction in proteotoxic and oxidative stress.
4.3. Energetic Preservation and Mitochondrial Efficiency
Our analysis of myocardial energetics revealed that CR preserved ATP content and ATP5G2 expression in NPR1 KO hearts. Interestingly, CR typically lowers ATP levels in WT hearts, a reflection of shifted metabolic demand, but in the KO heart, ATP levels remained relatively stable. This suggests that CR enhances mitochondrial efficiency or reduces metabolic demand independently of NPR1. However, the preservation of energy stores was not enough to rescue function, reinforcing the idea that the failure of the NPR1 KO heart is a signaling failure rather than a simple fuel shortage.
4.4. Impaired cGMP Generation as a Major Signaling Limitation
The profound reduction in myocardial cGMP levels (~90%) in NPR1 KO mice appears to represent a critical barrier to CR-mediated protection. In these hearts, we observed a compensatory upregulation of ANP and BNP at both mRNA and protein levels, accompanied by increased NO production and sGC expression (Figure 6). These responses suggest that the myocardium senses the loss of cGMP and attempts to compensate through enhanced ligand production and activation of the NO-sGC pathway. Despite these compensatory efforts, myocardial cGMP levels remained critically low and were not restored by CR.
At first glance, this finding may appear unexpected in light of prior studies demonstrating an essential role for eNOS/NO signaling in CR-mediated protection against ischemia-reperfusion cardiac injury [47] and ischemic limb muscle damage [48]. To reconcile our results with these reports, it’s possible that our analyses of whole-heart lysates may dilute endothelial eNOS-derived signals. Nevertheless, we observed consistent trends toward increased NO production in CR-treated WT hearts (Figure 6A,B), although these did not reach statistical significance; importantly, this should not be interpreted as a lack of biological relevance. Instead, our findings support the concept that NPR1-derived and NO-derived cGMP pools fulfill distinct and non-redundant roles in CR-induced cardioprotection, revealing a major signaling limitation at the level of NPR1-dependent cGMP generation.
Finally, although impaired NPR1-derived cGMP synthesis appears central, we cannot exclude additional contributing mechanisms, including enhanced cGMP degradation via PDE5 or PDE9, altered compartmentalized cGMP signaling, or reduced downstream PKG activation [49].
4.5. Translational Implications
In humans, NPR1 function varies widely due to genetic, metabolic, and environmental factors, and impaired natriuretic peptide signaling is common in obesity, hypertension, and HFpEF, conditions in which CR and dietary interventions are increasingly applied [15,21,22].
If intact NPR1 signaling is required for the full cardioprotective effects of CR, individuals with reduced NPR1 activity may derive diminished benefit, potentially contributing to heterogeneous responses observed in clinical lifestyle studies. These findings further suggest that pharmacologic enhancement of NPR1 signaling, such as neprilysin inhibition, designer natriuretic peptides, or NPR1 agonists, may synergize with CR to maximize cardiovascular benefit. Indeed, sacubitril/valsartan (Entresto) has demonstrated robust reductions in cardiovascular mortality and heart failure hospitalization in landmark trials, supporting the efficacy and safety of increasing ANP/BNP by neprilysin inhibition [50,51,52].
Finally, the inability of NO-sGC signaling to compensate for NPR1 loss underscores the specificity and nonredundancy of natriuretic peptide pathways in suppressing cardiac hypertrophy, indicating that therapeutic strategies should prioritize direct reinforcement of NPR1 signaling rather than relying solely on NO-based approaches.
4.6. Limitations of The Study
While our study provides evidence that NPR1 is essential for the cardioprotective effects of CR, several limitations should be considered when interpreting these findings. First, the CR protocol used here was relatively short-term (4 weeks). Longer CR interventions may elicit additional adaptations that could partially compensate for NPR1 deficiency. However, the profound remodeling observed in NPR1 KO mice suggests that even chronic CR would be insufficient without NPR1 signaling. Importantly, this study did not include NPR1 KO mice subjected to TAC under CR, which would be required to definitively test the necessity of NPR1 signaling in CR-mediated protection against acute pressure overload. Additionally, diastolic function was not assessed, and fractional shortening may not fully capture functional impairment, particularly in NPR1-deficient hearts. Negative findings in NPR1 KO mice should therefore be interpreted cautiously, as the study was not powered to detect small-to-moderate improvements in established diseases.
Second, this study employed global NPR1 knockout mice, which limits our ability to distinguish cardiomyocyte-specific effects from those arising in non-myocyte populations, such as cardiac fibroblasts. Future studies using cell type-specific NPR1 deletion models will be necessary to delineate the relative contributions of NPR1 signaling in cardiomyocytes versus fibroblasts during cardiac remodeling. Moreover, the use of a global knockout model precludes definitive separation of the direct cardiac effects of caloric restriction (CR) from its systemic actions on other organs. Because NPR1 is also expressed in the vasculature and kidneys, loss of NPR1 in these tissues may promote systemic hypertension or altered volume regulation, which likely contributes to the observed cardiac phenotype. Such systemic effects may, in turn, mask potential cell-autonomous protective actions of CR within cardiomyocytes themselves.
Third, while we measured total cellular cGMP, we did not assess subcellular cGMP pools using advanced imaging techniques. Such studies could confirm the compartmentalized nature of NPR1 versus sGC signaling [53].
Another important consideration in interpreting these findings is the fundamental difference between the pathological models examined. TAC in WT mice represents an acute hemodynamic stress imposed on a previously healthy heart, whereas NPR1 deficiency produces chronic, developmentally established cardiomyopathy with limited reversibility. As such, direct equivalence between these models should be avoided, and our conclusions are best interpreted as demonstrating the ability of CR to reduce acute cardiac remodeling where the NPR1 signaling is intact rather than an absolute requirement for all forms of cardioprotection.
Finally, the translational extrapolation to humans must be made cautiously. Human CR regimens are heterogeneous, and genetic variation in NPR1 expression or natriuretic peptide levels may modulate responses. Clinical studies incorporating biomarkers of NP-NPR1 activity will be necessary to validate our findings in patient populations.
5. Conclusions
Our findings identify the NPR1-cGMP signaling axis as a critical mediator of CR-associated protection against pressure-overload-induced cardiac remodeling. While CR activates broad metabolic and stress-resistance programs, these adaptations are insufficient to reverse established cardiomyopathy in the absence of functional NPR1. Collectively, these results suggest that effective cardioprotection requires intact NPR1-cGMP signaling and support therapeutic strategies that combine metabolic interventions with targeted restoration of cGMP signaling.
Author Contributions
Conceptualization and experimental design: K.C. and Q.L. Investigation: K.C., D.T. and Y.H. Data analysis: K.C., S.K. and Q.L. Manuscript writing and editing: K.C., D.T., S.K. and Q.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Center for Research Resources (5P20RR017662-10) and the National Institute of General Medical Sciences (8P20GM103455-10), National Institutes of Health.
Institutional Review Board Statement
All animal protocols conformed to the Public Health Service Guide for Care and Use of Laboratory Animals. These protocols were approved by the Institutional Animal Care and Use Committee at Sanford Research/University of South Dakota (Protocol Number: 43-11-14D; Approval date: 11/17/2011)
Informed Consent Statement
The study did not involve humans.
Data Availability Statement
Not applicable since the study did not report any data.
Acknowledgments
We thank the Physiology Core Facility at Sanford Research/USD for performing the echocardiographic measurements.
Conflicts of Interest
The authors declare no conflict of interest.
Use of AI and AI-Assisted Technologies
During the preparation of this work, the authors used Microsoft Copilot to improve the English clarity and editing of this manuscript. The authors take full responsibility for the content of the published article.
References
- 1.
Castello, L.; Maina, M.; Testa, G.; et al. Alternate-day fasting reverses the age-associated hypertrophy phenotype in rat heart by influencing the ERK and PI3K signaling pathways. Mech. Ageing Dev. 2011, 132, 305–314. https://doi.org/10.1016/j.mad.2011.06.006.
- 2.
Varady, K.A.; Bhutani, S.; Klempel, M.C.; et al. Alternate day fasting for weight loss in normal weight and overweight subjects: a randomized controlled trial. Nutr. J. 2013, 12, 146. https://doi.org/10.1186/1475-2891-12-146.
- 3.
Horne, B.D.; Muhlestein, J.B.; Anderson, J.L. Health effects of intermittent fasting: hormesis or harm? A systematic review. Am. J. Clin. Nutr. 2015, 102, 464–470. https://doi.org/10.3945/ajcn.115.109553.
- 4.
Martin, B.; Golden, E.; Egan, J.M.; et al. Reduced energy intake: the secret to a long and healthy life? IBS J. Sci. 2007, 2, 35–39.
- 5.
Masoro, E.J. Caloric restriction-induced life extension of rats and mice: A critique of proposed mechanisms. Biochim. Et Biophys. Acta 2009, 1790, 1040–1048. https://doi.org/10.1016/j.bbagen.2009.02.011.
- 6.
Weindruch, R. The retardation of aging by caloric restriction: studies in rodents and primates. Toxicol. Pathol. 1996, 24, 742–745.
- 7.
Longo, V.D.; Mattson, M.P. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. https://doi.org/10.1016/j.cmet.2013.12.008.
- 8.
Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2016, 39, 46–58. https://doi.org/10.1016/j.arr.2016.10.005.
- 9.
Nicoll, R.; Henein, M.Y. Caloric Restriction and Its Effect on Blood Pressure, Heart Rate Variability and Arterial Stiffness and Dilatation: A Review of the Evidence. Int. J. Mol. Sci. 2018, 19, 751. https://doi.org/10.3390/ijms19030751.
- 10.
Fontana, L.; Klein, S. Aging, adiposity, and calorie restriction. JAMA 2007, 297, 986–994. https://doi.org/10.1001/jama.297.9.986.
- 11.
Colman, R.J.; Anderson, R.M.; Johnson, S.C.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. https://doi.org/10.1126/science.1173635.
- 12.
Hofer, S.J.; Davinelli, S. Editorial: Dietary Strategies for Healthy Aging-Caloric Restriction and Beyond. Front. Nutr. 2022, 9, 866928. https://doi.org/10.3389/fnut.2022.866928.
- 13.
Lee, C.; Longo, V. Dietary restriction with and without caloric restriction for healthy aging. F1000Research 2016, 5, 117. https://doi.org/10.12688/f1000research.7136.1.
- 14.
Kirkham, A.A.; Beka, V.; Prado, C.M. The effect of caloric restriction on blood pressure and cardiovascular function: A systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2021, 40, 728–739. https://doi.org/10.1016/j.clnu.2020.06.029.
- 15.
Meyer, T.E.; Kovacs, S.J.; Ehsani, A.A.; et al. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J. Am. Coll. Cardiol. 2006, 47, 398–402. https://doi.org/10.1016/j.jacc.2005.08.069.
- 16.
Mitterberger, M.C.; Mattesich, M.; Zwerschke, W. Bariatric surgery and diet-induced long-term caloric restriction protect subcutaneous adipose-derived stromal/progenitor cells and prolong their life span in formerly obese humans. Exp. Gerontol. 2014, 56, 106–113. https://doi.org/10.1016/j.exger.2014.03.030.
- 17.
Spindler, S.R. Caloric restriction: from soup to nuts. Ageing Res. Rev. 2010, 9, 324–353. https://doi.org/10.1016/j.arr.2009.10.003.
- 18.
Raeini-Sarjaz, M.; Vanstone, C.A.; Papamandjaris, A.A.; et al. Comparison of the effect of dietary fat restriction with that of energy restriction on human lipid metabolism. Am. J. Clin. Nutr. 2001, 73, 262–267.
- 19.
Muthukumar, A.; Zaman, K.; Lawrence, R.; et al. Food restriction and fish oil suppress atherogenic risk factors in lupus-prone (NZB x NZW) F1 mice. J. Clin. Immunol. 2003, 23, 23–33.
- 20.
Mattson, M.P.; Wan, R. Beneficial effects of intermittent fasting and caloric restriction on the cardiovascular and cerebrovascular systems. J. Nutr. Biochem. 2005, 16, 129–137. https://doi.org/10.1016/j.jnutbio.2004.12.007.
- 21.
Viljanen, A.P.; Karmi, A.; Borra, R.; et al. Effect of caloric restriction on myocardial fatty acid uptake, left ventricular mass, and cardiac work in obese adults. Am. J. Cardiol. 2009, 103, 1721–1726. https://doi.org/10.1016/j.amjcard.2009.02.025.
- 22.
Riordan, M.M.; Weiss, E.P.; Meyer, T.E.; et al. The effects of caloric restriction- and exercise-induced weight loss on left ventricular diastolic function. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1174–H1182. https://doi.org/10.1152/ajpheart.01236.2007.
- 23.
Pamplona, R.; Portero-Otin, M.; Requena, J.; et al. Oxidative, glycoxidative and lipoxidative damage to rat heart mitochondrial proteins is lower after 4 months of caloric restriction than in age-matched controls. Mech. Ageing Dev. 2002, 123, 1437–1446.
- 24.
Levinson, D.F.; Holmans, P.A.; Laurent, C.; et al. No major schizophrenia locus detected on chromosome 1q in a large multicenter sample. Science 2002, 296, 739–741. https://doi.org/10.1126/science.1069914.
- 25.
Dhahbi, J.M.; Tsuchiya, T.; Kim, H.J.; et al. Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 218–231.
- 26.
Dolinsky, V.W.; Morton, J.S.; Oka, T.; et al. Calorie restriction prevents hypertension and cardiac hypertrophy in the spontaneously hypertensive rat. Hypertension 2010, 56, 412–421. https://doi.org/10.1161/hypertensionaha.110.154732.
- 27.
Wan, R.; Ahmet, I.; Brown, M.; et al. Cardioprotective effect of intermittent fasting is associated with an elevation of adiponectin levels in rats. J. Nutr. Biochem. 2010, 21, 413–417. https://doi.org/10.1016/j.jnutbio.2009.01.020.
- 28.
Shinmura, K.; Tamaki, K.; Bolli, R. Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. J. Mol. Cell. Cardiol. 2005, 39, 285–296. https://doi.org/10.1016/j.yjmcc.2005.03.010.
- 29.
Shinmura, K.; Tamaki, K.; Bolli, R. Impact of 6-mo caloric restriction on myocardial ischemic tolerance: possible involvement of nitric oxide-dependent increase in nuclear Sirt1. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2348–H2355.
- 30.
Shinmura, K.; Tamaki, K.; Saito, K.; et al. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation 2007, 116, 2809–2817. https://doi.org/10.1161/circulationaha.107.725697.
- 31.
Katare, R.G.; Kakinuma, Y.; Arikawa, M.; et al. Chronic intermittent fasting improves the survival following large myocardial ischemia by activation of BDNF/VEGF/PI3K signaling pathway. J. Mol. Cell. Cardiol. 2009, 46, 405–412. https://doi.org/10.1016/j.yjmcc.2008.10.027.
- 32.
Guo, Z.; Wang, M.; Ying, X.; et al. Caloric restriction increases the resistance of aged heart to myocardial ischemia/reperfusion injury via modulating AMPK-SIRT(1)-PGC(1a) energy metabolism pathway. Sci. Rep. 2023, 13, 2045. https://doi.org/10.1038/s41598-023-27611-6.
- 33.
Chen, K.; Kobayashi, S.; Xu, X.; et al. AMP activated protein kinase is indispensable for myocardial adaptation to caloric restriction in mice. PLoS ONE 2013, 8, e59682. https://doi.org/10.1371/journal.pone.0059682.
- 34.
Wei, Z.; Yang, B.; Wang, H.; et al. Caloric restriction, Sirtuins, and cardiovascular diseases. Chin. Med. J. 2024, 137, 921–935. https://doi.org/10.1097/CM9.0000000000003056.
- 35.
Sciarretta, S.; Forte, M.; Castoldi, F.; et al. Caloric restriction mimetics for the treatment of cardiovascular diseases. Cardiovasc. Res. 2021, 117, 1434–1449. https://doi.org/10.1093/cvr/cvaa297.
- 36.
Ding, K.; Gui, Y.; Hou, X.; et al. Transient Receptor Potential Channels, Natriuretic Peptides, and Angiotensin Receptor-Neprilysin Inhibitors in Patients With Heart Failure. Front. Cardiovasc. Med. 2022, 9, 904881. https://doi.org/10.3389/fcvm.2022.904881.
- 37.
Pandey, K.N.; Vellaichamy, E. Regulation of cardiac angiotensin-converting enzyme and angiotensin AT1 receptor gene expression in Npr1 gene-disrupted mice. Clin. Exp. Pharmacol. Physiol. 2010, 37, e70–e77. https://doi.org/10.1111/j.1440-1681.2009.05315.x.
- 38.
Oliver, P.M.; Fox, J.E.; Kim, R.; et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc. Natl. Acad. Sci. USA 1997, 94, 14730–14735.
- 39.
Subramanian, U.; Ramasamy, C.; Ramachandran, S.; et al. Genetic Disruption of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Triggers Differential Cardiac Fibrosis and Disorders in Male and Female Mutant Mice: Role of TGF-beta1/SMAD Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 11487. https://doi.org/10.3390/ijms231911487.
- 40.
Pandey, K.N. Genetic Ablation and Guanylyl Cyclase/Natriuretic Peptide Receptor-A: Impact on the Pathophysiology of Cardiovascular Dysfunction. Int. J. Mol. Sci. 2019, 20, 3946. https://doi.org/10.3390/ijms20163946.
- 41.
Vellaichamy, E.; Das, S.; Subramanian, U.; et al. Genetically altered mutant mouse models of guanylyl cyclase/natriuretic peptide receptor-A exhibit the cardiac expression of proinflammatory mediators in a gene-dose-dependent manner. Endocrinology 2014, 155, 1045–1056. https://doi.org/10.1210/en.2013-1416.
- 42.
Giovou, A.E.; Gladka, M.M.; Christoffels, V.M. The Impact of Natriuretic Peptides on Heart Development, Homeostasis, and Disease. Cells 2024, 13, 931. https://doi.org/10.3390/cells13110931.
- 43.
Choi, M.R.; Fernandez, B.E. Protective Renal Effects of Atrial Natriuretic Peptide: Where Are We Now? Front. Physiol. 2021, 12, 680213. https://doi.org/10.3389/fphys.2021.680213.
- 44.
Dessi-Fulgheri, P.; Sarzani, R.; Serenelli, M.; et al. Low calorie diet enhances renal, hemodynamic, and humoral effects of exogenous atrial natriuretic peptide in obese hypertensives. Hypertension 1999, 33, 658–662. https://doi.org/10.1161/01.hyp.33.2.658.
- 45.
Maoz, E.; Shamiss, A.; Peleg, E.; et al. The role of atrial natriuretic peptide in natriuresis of fasting. J. Hypertens. 1992, 10, 1041–1044.
- 46.
Carper, D.; Lac, M.; Coue, M.; et al. Loss of atrial natriuretic peptide signaling causes insulin resistance, mitochondrial dysfunction, and low endurance capacity. Sci. Adv. 2024, 10, eadl4374. https://doi.org/10.1126/sciadv.adl4374.
- 47.
Shinmura, K.; Tamaki, K.; Ito, K.; et al. Indispensable role of endothelial nitric oxide synthase in caloric restriction-induced cardioprotection against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H894–H903. https://doi.org/10.1152/ajpheart.00333.2014.
- 48.
Kondo, M.; Shibata, R.; Miura, R.; et al. Caloric restriction stimulates revascularization in response to ischemia via adiponectin-mediated activation of endothelial nitric-oxide synthase. J. Biol. Chem. 2009, 284, 1718–1724. https://doi.org/10.1074/jbc.M805301200.
- 49.
Fiedler, B.; Lohmann, S.M.; Smolenski, A.; et al. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 11363–11368. https://doi.org/10.1073/pnas.162100799.
- 50.
Kommu, S.; Berg, R.L. The Efficacy and Safety of Sacubitril/Valsartan Compared to Valsartan in Patients with Heart Failure and Mildly Reduced and Preserved Ejection Fractions: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Clin. Med. 2024, 13, 1572. https://doi.org/10.3390/jcm13061572.
- 51.
Charkviani, M.; Krisanapan, P.; Thongprayoon, C.; et al. Systematic Review of Cardiovascular Benefits and Safety of Sacubitril-Valsartan in End-Stage Kidney Disease. Kidney Int. Rep. 2024, 9, 39–51. https://doi.org/10.1016/j.ekir.2023.10.008.
- 52.
Grewal, P.K.; Abboud, A.; Myserlis, E.P.; et al. Sacubitril/Valsartan and Cognitive Outcomes in Heart Failure With Reduced Ejection Fraction. JACC Adv. 2023, 2, 100372. https://doi.org/10.1016/j.jacadv.2023.100372.
- 53.
Andressen, K.W.; Sedneva-Lugovets, D.; Ovesen, M.; et al. Regulation, effects and monitoring of compartmented cGMP signalling in cardiomyocytes-current status. Br. J. Pharmacol. 2025. https://doi.org/10.1111/bph.70144.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com