Polymeric organic semiconductors play a pivotal role in the development of new organic thermoelectric materials, sensing, and organic optoelectronics. Understanding the relation between their structure and properties is essential for designing the next generation of materials with improved features. Theoretical modelling based on first-principles calculations is a powerful aid for interpreting experimental structural and spectroscopic data of π-conjugated polymers, both in their pristine and doped states. Two approaches are currently available, i.e., oligomeric models and one-dimensional (1D) periodic crystal, each of one with its advantages and disadvantages. The combination of both allows to overcome the limitations of each. However, to do this, it is fundamental to systematically address the differences between these two approaches (molecular models vs. crystals). Here, we present a first systematic comparison between oligomeric and (1D) crystal models for describing the structural and spectroscopic properties of polyconjugated polymers. Using polythiophene (PT) and poly(3-hexylthiophene) (P3HT) as prototypical benchmark systems, we examine the relationship between finite-size oligomers and the corresponding infinite polymer treated as a one-dimensional (1D) crystal under periodic boundary conditions. Density functional theory (DFT) calculations are performed to examine the convergence of geometrical parameters and vibrational features (IR and Raman) as the oligomer length increases. The 1D crystal model is then evaluated as the limiting case of the oligomeric series. Its advantages in simplifying vibrational mode analysis are illustrated. This study establishes a methodological framework connecting molecular and periodic models, with direct relevance to the interpretation of experimental vibrational spectra of conducting polymers.
- Open Access
- Article
Bridging Molecular and Periodic Models: A Theoretical Study of the Spectroscopic Properties of Polythiophene †
Author Information
Received: 09 Dec 2025 | Revised: 15 Jan 2026 | Accepted: 21 Jan 2026 | Published: 07 Apr 2026
Abstract
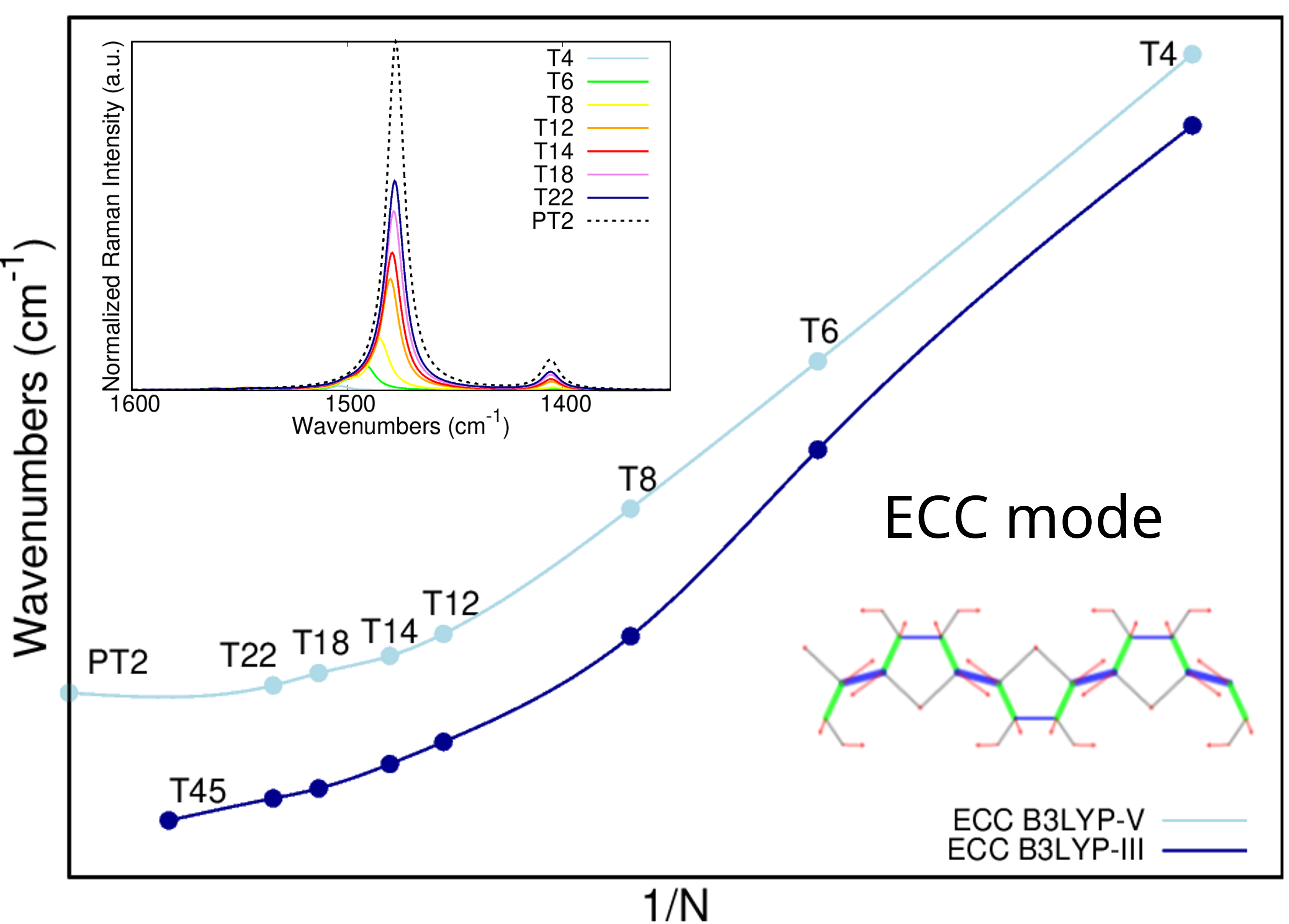
Graphical Abstract
Keywords
polythiophene | poly(3-hexylthiophene), infrared and Raman spectroscopy | conducting polymers | DFT calculations | periodic boundary conditions
References
- 1.
Skotheim, T.A.; Elsenbaumer, R.L.; Reynolds, J.R. (Eds.) Handbook of Conducting Polymers, 2nd ed.; Marcel Dekker: New York, NY, USA, 1988.
- 2.
Perepichka, J.F.; Perepichka, D.F. (Eds.) Handbook of Thiophene Based Materials; Wiley: Chichester, UK, 2009.
- 3.
Roth, S.; Carrol, D. One-Dimensional Metals; Wiley-VCH: Weinheim, Germany, 2004.
- 4.
Brédas, J.L.; Silbey, R. Conjugated Polymers; Springer: Dordrecht, The Netherlands, 1991.
- 5.
Pope, M.; Swenberg, C.E. Electronic Processes in Organic Crystals and Polymers; Oxford University Press: Oxford, UK, 1999.
- 6.
Kroon, R.; Mengistie, D.A.; Kiefer, D.; et al. Thermoelectric Plastics: From Design to Synthesis, Processing and Structure–Property Relationships. Chem. Soc. Rev. 2016, 45, 6147–6164.
- 7.
Kim, T.; Yang, S.J.; Sung, S.J.; et al. Highly Reproducible Thermocontrolled Electrospun Fiber Based Organic Photovoltaic Devices. ACS Appl. Mater. Interfaces 2015, 7, 4481–4487.
- 8.
Bharti, M.; Singh, A.; Samanta, S.; et al. Conductive Polymers for Thermoelectric Power Generation. Prog. Mater. Sci. 2018, 93, 270–310.
- 9.
Ludwigs, S. (Ed.) P3HT Revisited—From Molecular Scale to Solar Cell Devices; Springer: Berlin, Germany, 2014; Volume 256.
- 10.
Sumdani, G.M.; Islam, R.M.; Yahaya, N.A.A.; et al. Recent Advancements in Synthesis, Properties, and Applications of Conductive Polymers for Electrochemical Energy Storage Devices: A Review. Polym. Eng. Sci. 2022, 62, 269–303.
- 11.
Tashiro, K.; Kobayashi, M.; Kawai, T.; et al. Crystal Structural Change in Poly(3-Alkyl Thiophene)s Induced by Iodine Doping as Studied by an Organized Combination of X-ray Diffraction, Infrared/Raman Spectroscopy and Computer Simulation Techniques. Polymer 1997, 38, 2867–2879.
- 12.
Brinkmann, M. Structure and Morphology Control in Thin Films of Regioregular Poly(3-hexylthiophene). J. Polym. Sci. B 2011, 49, 1218–1233.
- 13.
Rahimi, K.; Botiz, I.; Stingelin, N.; et al. Controllable Processes for Generating Large Single Crystals of Poly(3-hexylthiophene). Angew. Chem. Int. Ed. 2012, 51, 11131–11135.
- 14.
Dudenko, D.; Kiersnowski, A.; Shu, J.; et al. A Strategy for Revealing the Packing in Semicrystalline p-Conjugated Polymers: Crystal Structure of Bulk Poly-3-hexyl-thiophene (P3HT). Angew. Chem. Int. Ed. 2012, 51, 11068–11072.
- 15.
Kayunkid, N.; Uttiya, S.; Brinkmann, M. Structural Model of Regioregular Poly(3-hexylthiophene) Obtained by Electron Diffraction Analysis. Macromolecules 2010, 43, 4961–4967.
- 16.
Hamidi-Sakr, A.; Biniek, L.; Bantignies, J.-L.; et al. A Versatile Method to Fabricate Highly In-plane Aligned Conducting Polymer Films with Anisotropic Charge Transport and Thermoelectric Properties: The Key Role of Alkyl Side Chain Layers on the Doping Mechanism. Adv. Funct. Mater. 2017, 27, 1700173.
- 17.
Lee, S.; Moon, G.D.; Jeong, U. Continuous Production of Uniform Poly(3-hexylthiophene) (P3HT) Nanofibers by Electrospinning and Their Electrical Properties. J. Mater. Chem. 2009, 19, 743–748.
- 18.
Chen, J.Y.; Kuo, C.C.; Lai, C.S.; et al. Manipulation on the Morphology and Electrical Properties of Aligned Electrospun Nanofibers of Poly(3-hexylthiophene) for Field-Effect Transistor Applications. Macromolecules 2011, 44, 2883–2892.
- 19.
Meng, B.; Liu, J.; Wang, L. Recent Development of n-Type Thermoelectric Materials Based on Conjugated Polymers. Nano Mat. Sci. 2021, 3, 113–123.
- 20.
Liu, X.; Yan, Y.; Zhang, Q.; et al. n-Type D-A Conjugated Polymers: Relationship Between Microstructure and Electrical/Mechanical Performance. Chem. Res. Chin. Univ. 2021, 37, 1019–1030.
- 21.
Du, C.H.; Xu, Y.H.; Li, H.; et al. Tough Hydrogen Bonding Crosslinked Poly(3-fluorothiophene) Network Via Electrosynthesis for High-Performance Electrochromic Supercapacitors. Chin. J. Polym. Sci. 2024, 42, 1749–1757.
- 22.
Wu, Z.; Zhao, Q.; Luo, X.; et al. Low-Cost Fabrication of High-Performance Fluorinated Polythiophene-Based Vis−NIR Electrochromic Devices Toward Deformable Display and Camouflage. Chem. Mater. 2022, 34, 9923−9933.
- 23.
Sugiyasu, K.; Honsho, Y.; Harrison, R.M.; et al. A Self-Threading Polythiophene: Defect-Free Insulated Molecular Wires Endowed with Long Effective Conjugation Length. J. Am. Chem. Soc. 2010, 132, 14754−14756.
- 24.
Ouchi, Y.; Sugiyasu, K.; Ogi, S.; et al. Synthesis of Self-Threading Bithiophenes and Their Structure-Property Relationships Regarding Cyclic Side-Chains with Atomic Precision. Chem. Asian J. 2012, 7, 75−84.
- 25.
Du, C.; Cheng, X.; Zhang, G.; et al. Molecular Bonding Engineering Enables Ultra-Stable Electrochromic Energy Storage in Flexible PEDOT Devices. Adv. Mater. 2025, e16740. https://doi.org/10.1002/adma.202516740
- 26.
Zbinden, R. Infrared Spectroscopy of High Polymers; Academic Press: New York, NY, USA, 1964.
- 27.
Painter, P.C.; Coleman, M.M.; Koening, J.L. The Theory of Vibrational Spectroscopy and Its Application to Polymeric Materials; Wiley: Chichester, UK, 1982.
- 28.
Zerbi, G. Molecular Vibrations of High Polymers. In Applied Spectroscopy Reviews, 2nd ed.; Brame, A.D., Ed.; Dekker: New York, NY, USA, 1969; Volume 2, p. 193.
- 29.
Zerbi, G. Vibrational Spectroscopy of Very Large Molecules. In Advances in Infrared and Raman Spectroscopy; Clark, R.J.H., Hester, R.E., Eds.; Heyden: London, UK, 1984; Volume 11, p. 301.
- 30.
Zerbi, G. Modern Polymer Spectroscopy; Wiley-VCH: New York, NY, USA, 1999; p. 113.
- 31.
Zerbi, G. Vibrational Spectroscopy of Polymers; Everall, N.J., Chalmers, J.M., Griffith, P.R., Eds.; Wiley: Chichester, UK, 2007; p. 487.
- 32.
Müllen, K.; Wegner, G. Electronic Materials: The Oligomer Approach; Wiley-VCH: Weinheim, Germany, 1998.
- 33.
Zerbi, G.; Chierichetti, B.; Ingänas, O. Vibrational Spectra of Oligothiophenes as Model of Polythiophenes. J. Chem. Phys. 1991, 94, 4637–4645.
- 34.
Zerbi, G.; Chierichetti, B.; Ingänas, O. Thermochromism in Polyalkylthiophenes: Molecular Aspects from Vibrational Spectroscopy. J. Chem. Phys. 1991, 94, 4646–4658.
- 35.
Agosti, E.; Rivola, M.; Hernandez, V.; et al. Electronic and Dynamical Effects from the Unusual Features of the Raman Spectra of Oligo and Polythiophenes. Synth. Met. 1999, 100, 101–112.
- 36.
Saporiti, C.; Brambilla, L.; Fazzi, D.; et al. Insights into the Structural and Vibrational Properties of Polaron in Doped Poly(3-alkyl-thiophene), P3HT. J. Phys. Chem. C. 2024, 128, 5189–5205.
- 37.
Anderson, M.; Ramanan, C.; Fontanesi, C.; et al. Displacement of Polarons by Vibrational Modes in Doped Conjugated Polymers. Phys. Rev. Mater. 2017, 1, 055604.
- 38.
Yin, J.; Wang, Z.; Fazzi, D.; et al. First-principles Study of the Nuclear Dynamics of Doped Conjugated Polymers. J. Phys. Chem. C 2016, 120, 1994–2001.
- 39.
Wang, S.; Fazzi, D.; Puttisong, Y.; et al. Effect of Backbone Regiochemistry on Conductivity, Charge Density, and Polaron Structure of n-Doped Donor–Acceptor Polymers. Chem. Mater. 2019, 31, 3395–3406.
- 40.
Furukawa, Y.; Akimoto, M.; Harada, I. Vibrational Key Bands and Electrical Conductivity of Polythiophene. Synth. Met. 1987, 18, 151–156.
- 41.
Cao, Y.; Renyuan, Q. IR and Raman Studies of Polythiophene Prepared by Electrochemical Polymerization. Solid State Commun. 1985, 54, 211–213.
- 42.
Louarn, G.; Trznadel, M.; Buisson, J.P.; et al. Raman Spectroscopic Studies of Regioregular Poly(3-alkylthiophenes). J. Phys. Chem. 1996, 100, 12532–12539.
- 43.
Brambilla, L.; Tommasini, M.; Botiz, I.; et al. Regio-Regular Oligo and Poly(3-HexylThiophene): Precise Structural Markers from the Vibrational Spectra of Oligomer Single Crystals. Macromolecules 2014, 47, 6730–6739.
- 44.
Brambilla, L.; Ferrón, C.C.; Tommasini, M.; et al. Infrared and Multi-Wavelength Raman Spectroscopy of Regio-regular P3HT and Its Deutero Derivatives. J. Raman Spectrosc. 2018, 49, 569–580.
- 45.
Brambilla, L.; Kim, J.-S.; Kim, B.J.; et al. Poly(3-hexylthiophene-2.5-diyl): Evidence of Different Polymer Chain Conformations in the Solid State from a Combined Study of Regioregularity Control and Raman Spectroscopy. J. Mol. Struct. 2020, 1221, 128882.
- 46.
Scholes, D.T.; Yee, P.Y.; Lindemuth, J.R.; et al. The Effects of Crystallinity on Charge Transport and the Structure of Sequentially Processed F4TCNQ-Doped Conjugated Polymer Films. Adv. Funct. Mater. 2017, 27, 1702654.
- 47.
Stanfield, D.A.; Wu, Y.; Tolbert, S.H.; et al. Controlling the Formation of Charge Transfer Complexes in Chemically Doped Semiconducting Polymers. Chem. Mater. 2021, 33, 2343–2356.
- 48.
Stanfield, D.A.; Mehmedović, Z.; Schwartz, B.J. Vibrational Stark Effect Mapping of Polaron Delocalization in Chemically Doped Conjugated Polymers. Chem. Mater. 2021, 33, 8489−8500.
- 49.
Arrigoni, A.; Brambilla, L.; Castiglioni, C.; et al. Conducting Electrospun Nanofibres: Monitoring of Iodine Doping of P3HT Through Infrared (IRAV) and Raman (RaAV) Polaron Spectroscopic Features. Nanomaterials 2022, 12, 4308.
- 50.
Spano, F.C.; Silva, C.H. Aggregate Behavior in Polymeric Semiconductors. Annu. Rev. Phys. Chem. 2014, 65, 477–500.
- 51.
Hu, K.; Doti, S.; Brambilla, L.; et al. Vibrational Properties of Doped P3HT Chains in Solution: Insight into the Doping Mechanism from Infrared IRAV and Raman RaAV Bands. Molecules 2025, 30, 1403.
- 52.
Lopez-Navarrete, J.T.; Zerbi, G. Lattice Dynamics and Vibrational Spectra of Polythiophene. I. Oligomers and Polymer. J. Chem. Phys. 1991, 94, 957–964.
- 53.
Lopez-Navarrete, J.T.; Zerbi, G. Lattice Dynamics and Vibrational Spectra of Polythiophene. II. Effective Coordinate Theory, Doping Induced, and Photoexcited Spectra. J. Chem. Phys. 1991, 94, 965–970.
- 54.
Baggioli, A.; Meille, S.V.; Raos, G.; et al. Intramolecular CH/π Interactions in Alkylaromatics: Monomer Conformations for Poly(3-alkylthiophene) Atomistic Models. Int. J. Quantum Chem. 2013, 113, 2154–2162.
- 55.
Su, W.P.; Schrieffer, J.R.; Heeger, A.J. Soliton Excitations in Polyacetylene. Phys. Rev. B 1980, 22, 2099–2111.
- 56.
Mele, E.J.; Rice, M.J. Vibrational Excitations of Charged Solitons. Phys. Rev. Lett. 1980, 45, 926–929.
- 57.
Ehrenfreund, E.; Vardeny, Z.; Brafman, O.; et al. Amplitude and Phase Modes in Trans Polyacetylene Resonant Raman Scattering and Induced Infrared Activity. Phys. Rev. B 1987, 36, 1535–1553.
- 58.
Vardeny, Z.; Ehrenfreund, E.; Brafman, O.; et al. Resonant Raman Scattering from Amplitude Modes in trans-(CH)x and -(CD)x. Phys. Rev. Lett. 1983, 51, 2326–2329.
- 59.
Painelli, A.; Girlando, A.; Del Freo, L.; et al. Infrared Intensity and Local Vibrations of Charged Solitons. Phys. Rev. B 1997, 56, 15100–15108.
- 60.
Jacobs, I.E.; Cendra, C.; Harrelson, T.F.; et al. Polymorphism Controls the Degree of Charge Transfer in a Molecularly Doped Semiconducting Polymer. Mater. Horiz., 2018, 5, 655–660.
- 61.
Casalegno, M.; Famulari, A.; Meille, S.V. Modeling of Poly(3-hexylthiophene) and Its Oligomer’s Structure and Thermal Behavior with Different Force Fields: Insights into the Phase Transitions of Semiconducting Polymers. Macromolecules 2022, 55, 2398–2412.
- 62.
Vilela Oliveira, D.; Peintinger, M.F.; Laun, J.; et al. BSSE-Correction Scheme for Consistent Gaussian Basis Sets of Double- and Triple-zeta Valence with Polarization Quality for Solid-State Calculations, J. Comp. Chem. 2019, 40, 2364–2376.
- 63.
Dovesi, R.; Orlando, R.; Erba, A.; et al. CRYSTAL14: A Program for the Ab Initio Investigation of Crystalline Solids. Int. J. Quantum Chem. 2014, 114, 1287–1317.
- 64.
Dovesi, R.; Erba, A.; Orlando, R.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360.
- 65.
Dovesi, R.; Erba, A.; Orlando, R.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2017.
- 66.
Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- 67.
Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211.
- 68.
Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys. 1993, 98, 5648–5652.
- 69.
Lee, C.; Yang, W.; Parr, R.G. Development of the Colle–Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B 1988, 37, 785–789.
- 70.
Gussoni, M.; Castiglioni, C.; Zerbi, G. Vibrational Spectroscopy of Polyconjugated Materials: Polyacetylene and Polyenes. In Spectroscopy of Advanced Materials; Clark, R.J.H., Hester, R.E., Eds.; Wiley: New York, NY, USA, 1991; pp. 251–353.
- 71.
Castiglioni, C.; Navarrete, J.T.L.; Zerbi, G.; et al. A Simple Interpretation of the Vibrational Spectra of Undoped, Doped and Photoexcited Polyacetylene: Amplitude Mode Theory in the GF Formalism. Solid State Commun. 1988, 65, 625–630.
- 72.
Zerbi, G.; Gussoni, M.; Castiglioni, C. Vibrational Spectroscopy of Polyconjugated Aromatic Materials with Electrical Non-Linear Properties. In Conjugated Polymers; Brédas, J.L., Silbey, R., Eds.; Springer: Dordrecht, The Netherlands, 1991; pp. 435–507.
- 73.
Castiglioni, C.; Del Zoppo, M.; Zerbi, G. Vibrational Raman Spectroscopy of Polyconjugated Organic Oligomers and Polymers. J. Raman Spectrosc. 1993, 24, 485–494.
- 74.
Hernandez, V.; Castiglioni, C.; Del Zoppo, M.; et al. Confinement Potential and π-Electron Delocalization in Polyconjugated Organic Materials. Phys. Rev. B 1994, 50, 9815–9823.
- 75.
Castiglioni, C.; Tommasini, M.; Zerbi, G. Raman Spectroscopy of Polyconjugated Molecules and Materials: Confinement Effect in One and Two Dimensions. Philos. Trans. R. Soc. A 2004, 362, 2425–2459.
- 76.
Tommasini, M.; Fazzi, D.; Milani, A.; et al. Intramolecular Vibrational Force Fields for Linear Carbon Chains Through an Adaptative Linear Scaling Scheme. J. Phys. Chem. A, 2007, 45, 11645–11651.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com