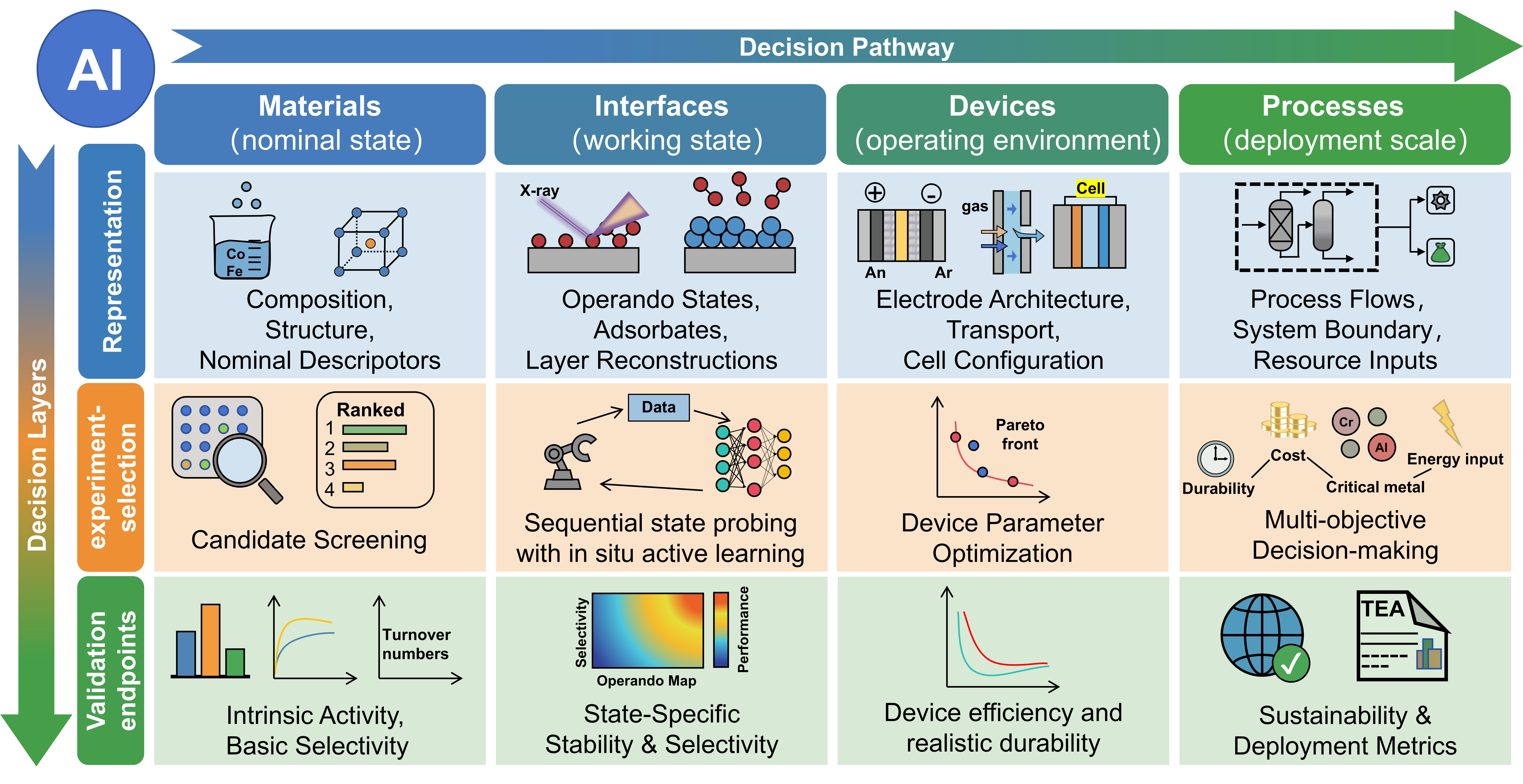
Machine learning is increasingly used to rank catalysts and accelerate discovery, but in energy catalysis the practical value of a model is rarely determined by ranking alone. Activity, selectivity and stability emerge from working catalysts and interfaces that depend on potential, solvent, temperature, pressure, transport and degradation, and these can differ substantially from the nominal materials used to build models. In this Review, we examine when screening-based workflows remain sufficient and when machine learning adds value beyond catalyst screening. We organize the field around three linked shifts, from static descriptors to operando-relevant representations, from offline prediction to closed-loop discovery, and from single-objective activity optimization to sustainability-aware multi-objective optimization. Using CO2 electroreduction, acidic oxygen evolution and higher alcohol synthesis as case studies, we show how these shifts can change catalyst ranking, experimental priorities and validation at the device or reactor level. We finally outline practical criteria for data reporting, extrapolative testing, uncertainty quantification and validation on the platform that defines success.
- Open Access
- Review
Artificial Intelligence in Energy Catalysis: From Catalyst Screening to Decision-Making
- Qixin Zhou 1,
- Yujie Mao 2,3,
- Wenjie Guo 4,
- Haifeng Shen 5,
- Haoying Wang 4,
- Yan Guo 2,*,
- Yongfa Zhu 4,*
Author Information
Received: 10 Apr 2026 | Revised: 08 May 2026 | Accepted: 15 May 2026 | Published: 18 May 2026
Abstract
Graphical Abstract
Keywords
artificial intelligence | energy catalysis | catalyst screening | operando catalysis | multi-objective optimization
References
- 1.
Kitchin, J.R. Machine Learning in Catalysis. Nat. Catal. 2018, 1, 230–232. https://doi.org/10.1038/s41929-018-0056-y.
- 2.
Li, Z.; Wang, S.; Xin, H. Toward Artificial Intelligence in Catalysis. Nat. Catal. 2018, 1, 641–642. https://doi.org/10.1038/s41929-018-0150-1.
- 3.
Freeze, J.G.; Kelly, H.R.; Batista, V.S. Search for Catalysts by Inverse Design. Artificial Intelligence, Mountain Climbers, and Alchemists. Chem. Rev. 2019, 119, 6595–6612. https://doi.org/10.1021/acs.chemrev.8b00759.
- 4.
Toyao, T.; Maeno, Z.; Takakusagi, S.; et al. Machine Learning for Catalysis Informatics. Recent Applications and Prospects. ACS Catal. 2020, 10, 2260–2297. https://doi.org/10.1021/acscatal.9b04186.
- 5.
Esterhuizen, J.A.; Goldsmith, B.R.; Linic, S. Interpretable Machine Learning for Knowledge Generation in Heterogeneous Catalysis. Nat. Catal. 2022, 5, 175–184. https://doi.org/10.1038/s41929-022-00744-z.
- 6.
Mou, L.-H.; Han, T.; Smith, P.E.S.; et al. Machine Learning Descriptors for Data‑Driven Catalysis Study. Adv. Sci. 2023, 10, 2301020. https://doi.org/10.1002/advs.202301020.
- 7.
Abraham, B.M.; Jyothirmai, M.V.; Sinha, P.; et al. Catalysis in the Digital Age. Unlocking the Power of Data with Machine Learning. WIREs Comput. Mol. Sci. 2024, 14, e1730. https://doi.org/10.1002/wcms.1730.
- 8.
Xin, H.; Kitchin, J.R.; López, N.; et al. Roadmap for Transforming Heterogeneous Catalysis with Artificial Intelligence. Nat. Catal. 2026, 9, 102–111. https://doi.org/10.1038/s41929-026-01479-x.
- 9.
Bozal-Ginesta, C.; Pablo-García, S.; Choi, C.; et al. Developing Machine Learning for Heterogeneous Catalysis with Experimental and Computational Data. Nat. Rev. Chem. 2025, 9, 601–616. https://doi.org/10.1038/s41570-025-00740-4.
- 10.
Zeng, Y.; Wang, J.; Li, F.; et al. AI-Accelerated Discovery of Electrocatalyst Materials. ACS Mater. Au 2026, 6, 72–89. https://doi.org/10.1021/acsmaterialsau.5c00135.
- 11.
Chen, H.; Kätelhön, E.; Lu, Y.; et al. 30 Years of AI for Electrocatalysis. Where We Are and Whatʼs Next. eScience 2026, 6, 100515. https://doi.org/10.1016/j.esci.2025.100515.
- 12.
Zhang, D.; Chen, Y.; Liu, C.; et al. Accelerating Catalyst Materials Discovery with Large Artificial Intelligence Models. Angew. Chem. Int. Ed. 2026, 65, e26150. https://doi.org/10.1002/anie.202526150.
- 13.
Zhang, D.; Jia, X.; Wang, Y.; et al. Digital Materials Ecosystem: From Databases to AI Agents for Autonomous Discovery. Chem. Sci. 2026, 17, 5782–5804. https://doi.org/10.1039/D5SC09229A.
- 14.
Liu, Z.; Liu, Y.; Zhang, Y.; et al. Universal Catalyst Design Framework for Electrochemical Hydrogen Peroxide Synthesis Facilitated by Local Atomic Environment Descriptors. Angew. Chem. Int. Ed. 2026, 65, e18027. https://doi.org/10.1002/anie.202518027.
- 15.
Wang, X.; Li, Z.; Zhang, D.; et al. Catalysis AI Agent Guides Discovering the Universal Design Principle of Cu-Based Single-Atom Alloy Catalysts for CO2 Electroreduction. Angew. Chem. Int. Ed. 2026, 65, e24612. https://doi.org/10.1002/anie.202524612.
- 16.
Prajapati, A.; Hahn, C.; Weidinger, I.M.; et al. Best Practices for in-Situ and Operando Techniques within Electrocatalytic Systems. Nat. Commun. 2025, 16, 2593. https://doi.org/10.1038/s41467-025-57563-6.
- 17.
Tian, X.; Tosello Gardini, A.; Raucci, U.; et al. Electrochemical Potential‑Driven Water Dynamics Control CO2 Electroreduction at the Ag-H2O Interface. Nat. Commun. 2025, 16, 10636. https://doi.org/10.1038/s41467-025-65630-1.
- 18.
Jia, X.; Wang, T.; Zhang, D.; et al. Advancing Electrocatalyst Discovery through the Lens of Data Science. State of the Art and Perspectives. J. Catal. 2025, 447, 116162. https://doi.org/10.1016/j.jcat.2025.116162.
- 19.
Savage, T.; Basha, N.; McDonough, J.; et al. Machine Learning‑Assisted Discovery of Flow Reactor Designs. Nat. Chem. Eng. 2024, 1, 522–531. https://doi.org/10.1038/s44286-024-00099-1.
- 20.
Peng, X.; Chen, L.; Liu, Y.; et al. Strain Engineering of Two‑Dimensional Materials for Energy Storage and Conversion Applications. Chem. Synth. 2023, 3, 47. https://doi.org/10.20517/cs.2023.34.
- 21.
Zhang, Y.; Liang, H. Crystal Facet Engineering of Electrocatalysts for Nitrate Reduction to Ammonia: Recent Advances and Future Perspectives. Chem. Synth. 2024, 4, 39. https://doi.org/10.20517/cs.2023.74.
- 22.
Xia, T.; Wang, X.; Wan, J.; et al. Asymmetrically Coordinated Single-Atom Catalysts: From Synthetic Strategy to Structure-Activity Relationship. Chem. Synth. 2025, 5, 74. https://doi.org/10.20517/cs.2025.08.
- 23.
Guo, Y.; Zhou, Q.; Nan, J.; et al. Perylenetetracarboxylic Acid Nanosheets with Internal Electric Fields and Anisotropic Charge Migration for Photocatalytic Hydrogen Evolution. Nat. Commun. 2022, 13, 2067. https://doi.org/10.1038/s41467-022-29826-z.
- 24.
Zhou, Q.; Guo, Y.; Zhu, Y. Photocatalytic Sacrificial H2 Evolution Dominated by Micropore-Confined Exciton Transfer in Hydrogen-Bonded Organic Frameworks. Nat. Catal. 2023, 6, 574–584. https://doi.org/10.1038/s41929-023-00972-x.
- 25.
Guo, Y.; Zhou, Q.; Wang, L.; et al. Enhanced Hydrogen Peroxide Photosynthesis via Charge-Complementary π-Electron Sites. Nat. Commun. 2025, 16, 6297. https://doi.org/10.1038/s41467-025-61452-3.
- 26.
Chai, H.; Guo, Y.; Nan, J.; et al. Proton-Conducting Hydrogen-Bonded Nanochannels Encapsulating Perylene-Based Photocatalysts for Solar Hydrogen Evolution. J. Mater. Chem. A 2026, 14, 10794–10799. https://doi.org/10.1039/D5TA10040B.
- 27.
Zhou, Q.; Guo, Y.; Zhu, Y. Reticular Copper Dual Sites Embedded with Semiconductor Particles for Selective CO2-to-C2H₄ Photoreduction. Nat. Catal. 2025, 8, 728–739. https://doi.org/10.1038/s41929-025-01369-8.
- 28.
Liu, W. Shining Light on Electrochemistry: A Synchrotron-Based X-ray Spectroscopic Interrogation. Chem. Synth. 2024, 4, 13. https://doi.org/10.20517/cs.2023.49.
- 29.
Sun, M.; Jin, B.; Yang, X.; et al. Probing Nuclear Quantum Effects in Electrocatalysis via a Machine-Learning-Enhanced Grand-Canonical Constant-Potential Approach. Nat. Commun. 2025, 16, 3600. https://doi.org/10.1038/s41467-025-58871-7.
- 30.
Sweeney, D.M.; Tran, B.; Goldsmith, B.R. A Grand-Canonical Study of the Potential Dependence of Nitrate Adsorption and Dissociation across Metals and Dilute Alloys. Commun. Chem. 2025, 8, 182. https://doi.org/10.1038/s42004-025-01579-y.
- 31.
Gauthier, A.; Vancauwenberghe, L.; Cousty, J.-C.; et al. A FAIR Research Data Infrastructure for High-Throughput Digital Chemistry. Digit. Discov. 2025, 4, 3502–3514. https://doi.org/10.1039/D5DD00297D.
- 32.
Mendes, P.S.F.; López, N.; De, S.; et al. Data as a Key Resource in Catalysis. A Community Account. ChemCatChem 2025, 17, e01226. https://doi.org/10.1002/cctc.202501226.
- 33.
Schumann, J.; Näsström, H.; Götte, M.; et al. Enabling Open and FAIR Catalysis Data with Standardized Data Structures. Nat. Catal. 2026, 9, 225–229. https://doi.org/10.1038/s41929-026-01508-9.
- 34.
Zheng, S.; Zhang, X.-M.; Liu, H.-S.; et al. Active Phase Discovery in Heterogeneous Catalysis via Topology-Guided Sampling and Machine Learning. Nat. Commun. 2025, 16, 2542. https://doi.org/10.1038/s41467-025-57824-4.
- 35.
Mom, R.V.; Falling, L.J.; Kasian, O.; et al. Operando Structure‑Activity‑Stability Relationship of Iridium Oxides during the Oxygen Evolution Reaction. ACS Catal. 2022, 12, 5174–5184. https://doi.org/10.1021/acscatal.1c05951.
- 36.
Ji, Q.; Tang, B.; Zhang, X.; et al. Operando Identification of the Oxide Path Mechanism with Different Dual-Active Sites for Acidic Water Oxidation. Nat. Commun. 2024, 15, 8089. https://doi.org/10.1038/s41467-024-52471-7.
- 37.
Baliyan, A.; Verma, S.; Sasakawa, K.; et al. Machine Learning-Guided Multimodal Synchrotron Analysis Workflow for Fuel Cell Electrocatalyst Discovery. Commun. Chem. 2025, 8, 376. https://doi.org/10.1038/s42004-025-01800-y.
- 38.
Lam, E.; Maury, T.; Preiss, S.; et al. General Data Management Workflow to Process Tabular Data in Automated and High-Throughput Heterogeneous Catalysis Research. Digit. Discov. 2025, 4, 539–547. https://doi.org/10.1039/D4DD00350K.
- 39.
Moshantaf, A.; Wesemann, M.; Beinlich, S.; et al. Advancing Catalysis Research through FAIR Data Principles Implemented in a Local Data Infrastructure. A Case Study of an Automated Test Reactor. Catal. Sci. Technol. 2024, 14, 6186–6197. https://doi.org/10.1039/D4CY00693C.
- 40.
Salim, I.; Berrouk, A.S. Toward Efficient Reporting for Catalytic Methane Reforming Data. A Framework and Review for Standardized Scientific Data Reporting. NPJ Comput. Mater. 2026, 12, 85. https://doi.org/10.1038/s41524-025-01953-3.
- 41.
Burte, A.S.; Nair, A.; Grabow, L.C.; et al. CatTestHub. A Benchmarking Database of Experimental Heterogeneous Catalysis for Evaluating Advanced Materials. J. Catal. 2025, 442, 115902. https://doi.org/10.1016/j.jcat.2024.115902.
- 42.
Wang, L.; Gao, Y.; Chen, X.; et al. A Corpus of CO2 Electrocatalytic Reduction Process Extracted from the Scientific Literature. Sci. Data 2023, 10, 175. https://doi.org/10.1038/s41597-023-02089-z.
- 43.
Chen, X.; Gao, Y.; Wang, L.; et al. Large Language Model Enhanced Corpus of CO2 Reduction Electrocatalysts and Synthesis Procedures. Sci. Data 2024, 11, 347. https://doi.org/10.1038/s41597-024-03180-9.
- 44.
Suvarna, M.; Zou, T.; Chong, S.H.; et al. Active Learning Streamlines Development of High Performance Catalysts for Higher Alcohol Synthesis. Nat. Commun. 2024, 15, 5844. https://doi.org/10.1038/s41467-024-50215-1.
- 45.
Ding, R.; Liu, J.; Hua, K.; et al. Leveraging Data Mining, Active Learning, and Domain Adaptation for Efficient Discovery of Advanced Oxygen Evolution Electrocatalysts. Sci. Adv. 2025, 11, eadr9038. https://doi.org/10.1126/sciadv.adr9038.
- 46.
Niu, X.; Chen, Y.; Sun, M.; et al. Bayesian Learning‑Assisted Catalyst Discovery for Efficient Iridium Utilization in Electrochemical Water Splitting. Sci. Adv. 2025, 11, eadw0894. https://doi.org/10.1126/sciadv.adw0894.
- 47.
Hung, L.; Yager, J.A.; Monteverde, D.; et al. Autonomous Laboratories for Accelerated Materials Discovery. A Community Survey and Practical Insights. Digit. Discov. 2024, 3, 1273–1279. https://doi.org/10.1039/D4DD00059E.
- 48.
Tom, G.; Schmid, S.P.; Baird, S.G.; et al. Self-Driving Laboratories for Chemistry and Materials Science. Chem. Rev. 2024, 124, 9633–9732. https://doi.org/10.1021/acs.chemrev.4c00055.
- 49.
Scheurer, C.; Reuter, K. Role of the Human-in-the-Loop in Emerging Self-Driving Laboratories for Heterogeneous Catalysis. Nat. Catal. 2025, 8, 13–19. https://doi.org/10.1038/s41929-024-01275-5.
- 50.
Volk, A.A.; Abolhasani, M. Performance Metrics to Unleash the Power of Self‑Driving Labs in Chemistry and Materials Science. Nat. Commun. 2024, 15, 1378. https://doi.org/10.1038/s41467-024-45569-5.
- 51.
Adesiji, A.D.; Wang, J.; Kuo, C.-S.; et al. Benchmarking Self-Driving Labs. Digit. Discov. 2026, 5, 14–27. https://doi.org/10.1039/D5DD00337G.
- 52.
Zhu, M.; Mroz, A.; Gui, L.; et al. Discrete and Mixed-Variable Experimental Design with Surrogate-Based Approach. Digit. Discov. 2024, 3, 2589–2606. https://doi.org/10.1039/D4DD00113C.
- 53.
Salazar-Villacis, P.; Benyahia, B. The ADePT Framework for Assessing Autonomous Laboratory Robotics. Commun. Chem. 2026, 9, 99. https://doi.org/10.1038/s42004-026-01932-9.
- 54.
Han, N.; Guo, H.; Zhang, B.; et al. Artificial Intelligence as a Driver of Sustainable Materials and Circularity. Nat. Rev. Mater. 2026. https://doi.org/10.1038/s41578-026-00912-8.
- 55.
Holman, P.A.; Shonnard, D.R.; Holles, J.H. Using Life Cycle Assessment to Guide Catalysis Research. Ind. Eng. Chem. Res. 2009, 48, 6668–6674. https://doi.org/10.1021/ie801934s.
- 56.
Artz, J.; Müller, T.E.; Thenert, K.; et al. Sustainable Conversion of Carbon Dioxide. An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. https://doi.org/10.1021/acs.chemrev.7b00435.
- 57.
Charalambous, C.; Moubarak, E.; Schilling, J.; et al. A Holistic Platform for Accelerating Sorbent‑Based Carbon Capture. Nature 2024, 632, 89–94. https://doi.org/10.1038/s41586-024-07683-8.
- 58.
Parveen, F.; Slater, A.G. Digitalisation of Catalytic Processes for Sustainable Production of Biobased Chemicals and Exploration of Wider Chemical Space. Catal. Sci. Technol. 2025, 15, 1689–1701. https://doi.org/10.1039/D4CY01525H.
- 59.
Wang, C.; Lai, H.; Warkentin, H.; et al. Catalysts for Electrochemical CO2 Conversion. Material Sustainability Perspective. NPJ Mater. Sustain. 2025, 3, 22. https://doi.org/10.1038/s44296-025-00065-9.
- 60.
Xun, D.; Liu, M.; Hao, H.; et al. Sustainable Supply of Critical Materials for Water Electrolysers and Fuel Cells. Commun. Earth Environ. 2025, 6, 627. https://doi.org/10.1038/s43247-025-02621-6.
- 61.
Chen, X.; El Khatib, M.; Lindgren, P.; et al. Atomistic Learning in the Electronically Grand-Canonical Ensemble. NPJ Comput. Mater. 2023, 9, 73. https://doi.org/10.1038/s41524-023-01007-6.
- 62.
Zhuang, Y.-B.; Liu, C.; Zhu, J.-X.; et al. An Artificial Intelligence Accelerated Ab Initio Molecular Dynamics Dataset for Electrochemical Interfaces. Sci. Data 2025, 12, 997. https://doi.org/10.1038/s41597-025-05338-5.
- 63.
Erlebach, A.; Šípka, M.; Saha, I.; et al. A Reactive Neural Network Framework for Water-Loaded Acidic Zeolites. Nat. Commun. 2024, 15, 4215. https://doi.org/10.1038/s41467-024-48609-2.
- 64.
Omranpour, A.; Elsner, J.; Lausch, K.N.; et al. Machine Learning Potentials for Heterogeneous Catalysis. ACS Catal. 2025, 15, 1616–1634. https://doi.org/10.1021/acscatal.4c06717.
- 65.
Tinajero, C.; Zanatta, M.; Sánchez-Velandia, J.E.; et al. Reac-Discovery. An Artificial Intelligence‑Driven Platform for Continuous-Flow Catalytic Reactor Discovery and Optimization. Nat. Commun. 2025, 16, 9062. https://doi.org/10.1038/s41467-025-64127-1.
- 66.
Bennett, J.A.; Orouji, N.; Khan, M.B.; et al. Autonomous Reaction Pareto-Front Mapping with a Self-Driving Catalysis Laboratory. Nat. Chem. Eng. 2024, 1, 240–250. https://doi.org/10.1038/s44286-024-00033-5.
- 67.
Zhang, C.; Lu, R.; Sun, Q.; et al. Finding the Pareto Front for High-Entropy-Alloy Catalysts. Chem. Sci. 2026, 17, 4744–4752. https://doi.org/10.1039/D5SC06100H.
- 68.
Kim, J.; Kwon, I.S.; Lim, J.; et al. Machine-Learning-Guided Tungsten Single Atoms Promote Oxyhydroxides for Noble-Metal-Free Water Electrolysis. Nat. Commun. 2026, 17, 2344. https://doi.org/10.1038/s41467-026-68735-3.
- 69.
Casillo, E.; Scattolin, T.; Nolan, S.P. Catalysis Meets Machine Learning. A Guide to Data-Driven Discovery and Design. Chem. Commun. 2025, 61, 18247–18272. https://doi.org/10.1039/D5CC05274B.
- 70.
Tran, K.; Ulissi, Z.W. Active Learning across Intermetallics to Guide Discovery of Electrocatalysts for CO2 Reduction and H2 Evolution. Nat. Catal. 2018, 1, 696–703. https://doi.org/10.1038/s41929-018-0142-1.
- 71.
Mok, D.H.; Li, H.; Zhang, G.; et al. Data-Driven Discovery of Electrocatalysts for CO2 Reduction Using Active Motifs‑Based Machine Learning. Nat. Commun. 2023, 14, 7303. https://doi.org/10.1038/s41467-023-43118-0.
- 72.
Wang, G.; Mine, S.; Chen, D.; et al. Accelerated Discovery of Multi-Elemental Reverse Water-Gas Shift Catalysts Using Extrapolative Machine Learning Approach. Nat. Commun. 2023, 14, 5861. https://doi.org/10.1038/s41467-023-41341-3.
- 73.
Zhong, M.; Tran, K.; Min, Y.; et al. Accelerated Discovery of CO2 Electrocatalysts Using Active Machine Learning. Nature 2020, 581, 178–183. https://doi.org/10.1038/s41586-020-2242-8.
- 74.
Song, Z.; Fan, L.; Lu, S.; et al. Inverse Design of Promising Electrocatalysts for CO2 Reduction via Generative Models and Bird Swarm Algorithm. Nat. Commun. 2025, 16, 1053. https://doi.org/10.1038/s41467-024-55613-z.
- 75.
Cui, Z.; Aztergo, K.D.; Hwang, J.; et al. Determining CO Adsorption Free Energies on CO2 Electroreduction Active Sites through Kinetic Analysis. Nat. Catal. 2025, 8, 1117–1127. https://doi.org/10.1038/s41929-025-01427-1.
- 76.
Zhan, C.; Dattila, F.; Rettenmaier, C.; et al. Key Intermediates and Cu Active Sites for CO2 Electroreduction to Ethylene and Ethanol. Nat. Energy 2024, 9, 1485–1496. https://doi.org/10.1038/s41560-024-01633-4.
- 77.
Zhang, Z.; Li, H.; Shao, Y.; et al. Molecular Understanding of the Critical Role of Alkali Metal Cations in Initiating CO2 Electroreduction on Cu(100) Surface. Nat. Commun. 2024, 15, 612. https://doi.org/10.1038/s41467-024-44896-x.
- 78.
Chen, J.; Qiu, H.; Zhao, Y.; et al. Selective and Stable CO2 Electroreduction at High Rates via Control of Local H2O to CO2 Ratio. Nat. Commun. 2024, 15, 5893. https://doi.org/10.1038/s41467-024-50269-1.
- 79.
Gao, X.; Jiang, Y.; Liu, J.; et al. Intermediate‑Regulated Dynamic Restructuring at Ag‑Cu Biphasic Interface Enables Selective CO2 Electroreduction to C2+ Fuels. Nat. Commun. 2024, 15, 10331. https://doi.org/10.1038/s41467-024-54630-2.
- 80.
Cheng, D.; Nguyen, K.L.C.; Sumaria, V.; et al. Structure Sensitivity and Catalyst Restructuring for CO2 Electro‑Reduction on Copper. Nat. Commun. 2025, 16, 4064. https://doi.org/10.1038/s41467-025-59267-3.
- 81.
Knöppel, J.; Möckl, M.; Escalera-López, D.; et al. On the Limitations in Assessing Stability of Oxygen Evolution Catalysts Using Aqueous Model Electrochemical Cells. Nat. Commun. 2021, 12, 2231. https://doi.org/10.1038/s41467-021-22296-9.
- 82.
Tao, H.B.; Liu, H.; Lao, K.; et al. The Gap between Academic Research on Proton Exchange Membrane Water Electrolysers and Industrial Demands. Nat. Nanotechnol. 2024, 19, 1074–1076. https://doi.org/10.1038/s41565-024-01699-x.
- 83.
Wang, C.R.; Stansberry, J.M.; Mukundan, R.; et al. Proton Exchange Membrane Water Electrolysis. Cell-Level Considerations for Gigawatt-Scale Deployment. Chem. Rev. 2025, 125, 1257–1302. https://doi.org/10.1021/acs.chemrev.3c00904.
- 84.
Park, W.; Noh, J.; Gu, G.H.; et al. Machine Learning-Enabled Fast Exploration of Stable and Active Single-Atom Catalysts for Oxygen Evolution Reaction. Innov. Mater. 2024, 2, 100072. https://doi.org/10.59717/j.xinn-mater.2024.100072.
- 85.
Abed, J.; Heras-Domingo, J.; Sanspeur, R.Y.; et al. Pourbaix Machine Learning Framework Identifies Acidic Water Oxidation Catalysts Exhibiting Suppressed Ruthenium Dissolution. J. Am. Chem. Soc. 2024, 146, 15740–15750. https://doi.org/10.1021/jacs.4c01353.
- 86.
Wu, Z.-Y.; Chen, F.-Y.; Li, B.; et al. Non-Iridium-Based Electrocatalyst for Durable Acidic Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysis. Nat. Mater. 2023, 22, 100–108. https://doi.org/10.1038/s41563-022-01380-5.
- 87.
Cao, X.; Miao, L.; Jia, W.; et al. Strain Heterogeneity in RuO2 for Efficient Acidic Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysis. Nat. Commun. 2025, 16, 6217. https://doi.org/10.1038/s41467-025-58570-3.
- 88.
Qian, F.; Cao, D.; Chen, S.; et al. High-Entropy RuO2 Catalyst with Dual-Site Oxide Path for Durable Acidic Oxygen Evolution Reaction. Nat. Commun. 2025, 16, 6894. https://doi.org/10.1038/s41467-025-61763-5.
- 89.
Zhang, K.; Liang, X.; Wang, Y.; et al. Support-Tuned Iridium Reconstruction with Crystalline Phase Dominating Acidic Oxygen Evolution. Nat. Commun. 2025, 16, 8164. https://doi.org/10.1038/s41467-025-63541-9.
- 90.
Yang, D.; Zhang, C.; Qin, Y.; et al. Single-Faceted IrO2 Monolayer Enabling High‑Performing Proton Exchange Membrane Water Electrolysis beyond 10,000 h Stability at 1.5 A cm−2. Nat. Commun. 2025, 16, 7236. https://doi.org/10.1038/s41467-025-62665-2.
- 91.
Qiu, C.; Sellers, C.; Wu, Z.-Y.; et al. Low‑Iridium Stabilized Ruthenium Oxide Anode Catalyst for Durable Proton-Exchange Membrane Water Electrolysis. Nat. Nanotechnol. 2025, 20, 1787–1795. https://doi.org/10.1038/s41565-025-02030-y.
- 92.
Suvarna, M.; Preikschas, P.; Pérez-Ramírez, J. Identifying Descriptors for Promoted Rhodium-Based Catalysts for Higher Alcohol Synthesis via Machine Learning. ACS Catal. 2022, 12, 15373–15385. https://doi.org/10.1021/acscatal.2c04349.
- 93.
Pisal, P.; Krejčí, O.; Rinke, P. Machine Learning Accelerated Descriptor Design for Catalyst Discovery in CO2 to Methanol Conversion. NPJ Comput. Mater. 2025, 11, 213. https://doi.org/10.1038/s41524-025-01664-9.
- 94.
Ramirez, A.; Lam, E.; Gutierrez, D.P.; et al. Accelerated Exploration of Heterogeneous CO2 Hydrogenation Catalysts by Bayesian-Optimized High-Throughput and Automated Experimentation. Chem Catal. 2024, 4, 100888. https://doi.org/10.1016/j.checat.2023.100888.
- 95.
Ge, Y.; Martín, A.J.; Suvarna, M.; et al. Descriptors toward Higher Alcohols and Olefins Formation in CO2 Hydrogenation on Promoted Iron Catalysts. Appl. Catal. B Environ. Energy 2025, 378, 125578. https://doi.org/10.1016/j.apcatb.2025.125578.
- 96.
Grasselli, F.; Chong, S.; Kapil, V.; et al. Uncertainty in the Era of Machine Learning for Atomistic Modeling. Digit. Discov. 2025, 4, 2654–2675. https://doi.org/10.1039/D5DD00102A.
- 97.
Bilbrey, J.A.; Firoz, J.S.; Lee, M.-S.; et al. Uncertainty Quantification for Neural Network Potential Foundation Models. NPJ Comput. Mater. 2025, 11, 109. https://doi.org/10.1038/s41524-025-01572-y.
- 98.
Nair, A.S.; Foppa, L.; Scheffler, M. Materials-Discovery Workflow Guided by Symbolic Regression for Identifying Acid-Stable Oxides for Electrocatalysis. NPJ Comput. Mater. 2025, 11, 150. https://doi.org/10.1038/s41524-025-01596-4.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com