

Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Recent Advances in Natural Plant-based Treatment of Myocardial Ischemia-reperfusion Injury
Peixun Yang 1,3,4, Minxuan Liu 2,3,4, Xiaoxue Fan 3,4, Xinzhuang Zhang 3,4, Liang Cao 3,4, Zhenzhong Wang 3,4, and Wei Xiao 3,4, *
1 Kanion School of Chinese Materia Medica, Nanjing University of Chinese Medicine, 138 Xianlin Avenue Qixia District, Nanjing 210046, China
2 School of Pharmacy, Nanjing University of Chinese Medicine, 138 Xianlin Avenue Qixia District, Nanjing 210046, China
3 National Key Laboratory on Technologies for Chinese Medicine Pharmaceutical Process Control and Intelligent Manufacture, Jiangning Industrial City, Economic and Technological Development Zone of Lianyungang, Lianyungang 222001, China
4 Jiangsu Kanion Pharmaceutical Co Ltd, Jiangning Industrial City, Economic and Technological Development Zone of Lianyungang, Lianyungang 222001, China
* Correspondence: xw_kanion@163.com ( Wei Xiao)
Received: 23 March 2023
Accepted: 10 May 2023
Published: 27 June 2023
Abstract: Cardiovascular disease (CDV) is the primary cause of death in the world, and myocardial ischemia (MI) is one of the high-risk CVDs. The myocardial blood supply must be restored as soon as possible to reduce the mortality risk, however, reperfusion itself paradoxically leads to further death of cardiomyocytes and increases the infarct size; this is known as myocardial ischemia/reperfusion injury (MIRI). The pathological mechanism of MIRI is complex, and current research mainly focuses on oxidative stress, dysfunctional mitochondrial energy metabolism, Ca 2+ overload, endoplasmic reticulum stress (ERs) and the inflammatory response. This review briefly summarizes the mechanism of MIRI, and natural plant product (NPP) components proven to ameliorate MIRI and their related signaling pathways. NPPs can alleviate MIRI by regulating oxidative stress, inflammation, ERs, Ca 2+ overload and mitochondrial function maintenance. This review will deepen our understanding of how NPPs reduce MIRI and the future value of NPPs in cardio-protection.
Keywords:
myocardial ischemia/reperfusion injury natural plant products mechanism signaling pathways1. Introduction
Cardiovascular disease is the leading cause of death all over the world; in 2019 it accounted for 18.6 million deaths, including 9.14 million deaths due to myocardial ischemia [1, 2]. In clinical settings, thrombolytic drugs and revascularization surgery are used to restore the blood supply of ischemic myocardium [3, 4]. However, the reperfusion paradoxically causes further cellular metabolic dysfunction, structural tissue injury, and even irreversible injury; this phenomenon is known as myocardial ischemia/reperfusion injury (MIRI). MIRI directly reduces the therapeutic effect of reperfusion and greatly increases the patient follow-up costs [5]. The pathological mechanism of MIRI is complex; it is associated with oxidative stress, mitochondrial dysfunction, Ca 2+ overload and inflammation [6]. These factors cause damage to the myocardial intima, myocardial contractile dysfunction, and cardiomyocyte death [7]. Clinical treatment of MIRI is mainly focused on inhibiting the inflammatory response, reducing oxidative stress, preventing Ca 2+ overload, and restoring energy metabolism [8-10].
Many natural medicines, especially natural plant products (NPPs), have multiple targets and low toxicity [11]. At present, a number of natural medicines have been found to exhibit therapeutic effects on MIRI, including glycosides, flavonoids, alkaloids, phenols, carbohydrates, terpenes, organic acids, amino acids and catechins. These natural substances play a vital role in decreasing MIRI by inhibiting oxidative stress and apoptosis, reducing mitochondrial damage, and improving energy metabolism [9]. This article summarizes the latest papers on the treatment of MIRI with NPPs, and discusses the intervention effect of NPPs on MIRI through inhibition of Ca 2+ overload, oxidative stress injury, mitochondrial damage, inflammatory response and other related mechanisms.
2. Mechanism of Myocardial Ischemia-reperfusion Injury
The mechanism of MIRI is related to oxidative stress, Ca 2+ overload, energy metabolism disorder, inflammatory response, endoplasmic reticulum (ER) stress, apoptosis and autophagy. During ischemia, decreased oxygen and nutrient supply leads to energy metabolism dysfunction, resulting in the accumulation of reactive oxygen species (ROS) and Ca 2+ overload, which can cause damage to cellular structures and organelles, such as mitochondria. When reperfusion begins, the sudden reintroduction of oxygen causes a surge in ROS and inflammatory responses, thereby exacerbating the MIRI. In addition, reperfusion injury impairs mitochondrial function, perpetuates ER stress, and leads to apoptosis and excessive autophagy. Various other factors also interact with each other in the process of MIRI.
2.1. Oxidative Stress
Under physiological conditions, the oxidants and the antioxidants system maintain a dynamic balance [12]. Ischemia/Reperfusion (I/R) progress produces excessive oxidants that break the balance between oxidants and the antioxidants. This phenomenon is called oxidative stress [13].
Elevated intracellular ROS level is an important characteristic of oxidative stress [14]. After the ischemic myocardium restores blood flow, oxygen molecules will produce a large amount of O2- under the catalysis of mitochondrial respiratory chain complex I and III, NADPH oxidase (NOX) and xanthine oxidases [12, 15, 16]. O2- is converted to H 2O 2 under the catalytic action of superoxide dismutase (SOD), and a large amount of H 2O 2 is also produced by the catalytic action of p66shc and monoamine oxidase isoforms (MAOs) [17, 18]. Due to large quantities, H2O2 cannot be completely detoxified by the peroxiredoxin/thioredoxin (PRX/Trx) system and glutathione peroxidase/reductase (GSH-PX) system; some H2O2 is converted to ·OH under the catalysis of Fe2+ or to hypochlorite (HClO) by the catalytic action of myeloperoxidase [17, 19].
ROS can directly damage the phospholipid bilayer of a cell membrane, leading to the imbalance of membrane components. Membrane fluidity and permeability are affected, which results in irreversible damage to organelles and cells [20]. Increase of ROS affects sarcoplasmic reticulum (SR), Ca2+-ATPase (SERCA2a) and calcium-sensing receptor (CaSR), resulting in Ca2+ overload and myocardial contractile dysfunction [21]. Oxidative stress also causes the continuous opening of mitochondrial permeability transition pore (MPTP) during reperfusion, and the destruction of MPTP leads to mitochondrial rupture and damage, which affects cell energy metabolism [22]. ROS causes damage to DNA, which affects the correct synthesis of proteins, leading to cell death. Oxidative stress also causes an inflammatory reaction cascade; inflammatory factors are released and further reperfusion injury occurs [13].
Recent articles showed that NPPs could reduce MIRI by regulating related pathway proteins. For example, luteolin inhibits oxidative stress by inhibiting the mitogen-activated protein kinase (MAPK) pathway, and dihydroquercetin inhibits oxidative stress and reduces cardiomyocyte apoptosis by activating phosphoinositide-3 kinase (PI3K)/protein kinase B (AKT) pathway [23, 24].
2.2. Ca 2+ Overload
Ca2+ is mainly distributed in the mitochondria and the sarcoplasmic reticulum (SR) of the cell, and it is an important signaling molecule in excitation–contraction coupling (ECC) [25]. During myocardial ischemia, reduced synthesis of adenosine-triphosphate (ATP) decreases the activity of SERCA2a and intracellular Ca2+ accumulates, resulting in intracellular Ca2+ overload [26, 27]. At the same time, during the ischemic phase, a large amount of H+ is produced in the cell due to anaerobic respiration, resulting in a large amount of Na+ entering the cell through H+/Na+ exchange (HNX) [28]. The excessive amount of intracellular Na+ increases the activity of Na+/Ca2+ exchange (NCX) proteins, which promotes Na+ excretion and Ca2+ intake. The increased intracellular Ca2+ concentration leads to Ca2+ overload [29]. When reperfusion occurs, the extracellular pH rises rapidly, further increasing HNX and NCX activity and exacerbating intracellular calcium overload [30]. Due to the presence of Ca2+ in the cytoplasm, the opening of ryanodine receptor 2 (RyR2) channels in the SR leads to Ca2+ influx from the SR into the cytoplasm, which also exacerbates intracellular Ca2+ overload [8]. Moreover, a large amount of ROS is generated during reperfusion, which damages the cell membrane. Altered cell membrane permeability then causes inward flow of Ca2+ and ER damage, which exacerbates Ca2+ overload [31].
During MIRI, a lot of Ca2+ accumulates in the cytoplasm. To maintain cytoplasmic Ca 2+ homeostasis, mitochondria will take up excessive Ca2+ in the cytoplasm. This Ca2+ overload causes changes in mitochondrial membrane potential (MMP) and mitochondrial morphology [32, 33]. Moreover, excessive Ca2+ in mitochondria inhibits ATP synthesis and disturbs mitochondrial energy metabolism [6, 25]. Intracellular Ca2+ also activates protein kinase A/phospholipase A, causing the breakdown of membrane phospholipids into free fatty acids, leukotrienes, ROS and other toxic substances, which causes changes in membrane permeability. Toxic substances also induce energy metabolism disorders and cell signaling, promoting apoptosis [34, 35]. Furthermore, Ca2+ overload causes structural and functional changes in coronary macrovascular and microvascular endothelial cells. These changes alter the vascular permeability, causing neutrophil adhesion, accumulation and infiltration, which release a series of inflammatory mediators that further damage the cardiovascular tissue [36, 37].
Due to the close connection between calcium-related channel proteins and the apoptotic pathway, many NPPs inhibit Ca2+ overload by regulating Ca 2+ channel protein-mediated apoptosis pathways, such as Astragaloside IV (AsⅣ), Hydroxysafflor Yellow A (HSYA) and total saponins of Aralia Elata [38-40].
2.3. Mitochondrial Dysfunction
Mitochondria is an important organelle that provides ATP and maintains energy metabolism [41]. The energy metabolism disorder caused by mitochondrial damage during reperfusion is an important mechanism leading to the death of cardiomyocytes [42]. MPTP opening is triggered by the anaerobic respiration, damage to the mitochondrial electron transport chain (ETC) during ischemia, mitochondrial Ca 2+ overload, and excessive ROS [17, 43]. The opening of MPTP leads to mitochondrial swelling and MMP loss [25, 44].
During I/R, dysfunction in mitochondrial dynamics is a considerable cause of cardiac injury [45]. Under normal conditions, mitochondria fission and fusion depend on energy needs [46]. After I/R, the content of Mitofusion2 (Mfn2) in cardiomyocytes is decreased, while the content of dynamin-related protein 1(Drp1) is increased and translocated to the mitochondrial membrane, resulting in increased mitochondrial fission and decreased fusion [42]. The opening of MPTP not only reduces production of ATP, but also promotes the release of cytochrome c (Cyt c) and causes myocardial cell apoptosis [47]. Excessive fission leads to mutations in mitochondrial DNA fragments, aggravates Ca2+ overload and ROS burst [46].
Moderate mitophagy can eliminate damaged mitochondria and maintain mitochondrial quality, which is a significant way to reduce I/R [42]. During I/R, the depolarization of MMP causes the inhibition of the degradation process of PTEN-induced putative kinase 1 (PINK1), resulting in the accumulation of PINK1 on mitochondrial membranes [48, 49]. Parkin is recruited to the mitochondria and attached to the site of ubiquitin protein through PINK1 on the mitochondrial membrane or through a PINK1-independent mechanism. The ubiquitinated mitochondrial protein is recognized by protein p62/sequestosome 1 and then linked to MAP1LC3 (LC3) on autophagosomes to engulf damaged mitochondria [50].
During the I/R process, the function, morphology, and dynamics of mitochondria are affected, resulting in cell apoptosis [42]. Mitochondrial damage is the result of multiple mechanisms, so there are many different therapeutic strategies for drugs that target mitochondria. Therefore, modulation of the damaged mitochondria during I/R includes modulation of electron transport, mitophagy and mitochondrial dynamic changes, inhibition of MPTP opening, Ca 2+ overload and oxidative stress. For example, sappanone is a post-conditioning substance, which can reduce myocardial dysfunction and injury by regulating MPTP opening, mitophagy and mitochondrial dynamic via AMPK activation [51]. Moreover, Icariin could alleviate mitochondrial oxidative stress and apoptosis via SIRT1-dependent mechanism [52].
2.4. Endoplasmic Reticulum Stress
Endoplasmic reticulum (ER) is a very important organelle in eukaryotic cells; it maintains intracellular Ca2+ homeostasis, and synthesizes cholesterol and lipids. ER is also essential for protein synthesis, transport and modification [53, 54]. In physiological conditions, ER quality control system (ERQC) promotes the correct folding of proteins and degradation of misfolded peptides, which is regulated by folding enzymes, lectins and molecular chaperones [55]. During I/R, Ca 2+ overload, oxidative stress and other factors will lead to ERQC dysfunction, causing the wrong proteins to accumulate, and eventually resulting in ER stress [56, 57]. In the early stage of ER stress, many chaperone genes are induced at the transcriptional level to clear accumulated unfolded proteins in time and to activate the ER-associated degradation (ERAD) system, which can send misfolded peptides to the cytoplasm for proteasome degradation. These processes are named unfolded protein response (UPR) [53, 58].
UPR is regulated by a chaperone immunoglobulin/78 kDa glucose regulatory protein (BIP/GRP78) and three transmembrane signal sensors, namely, protein kinase-like ER kinase (PERK), activation of transcription factor 6 (ATF6), and inositol-requiring kinase 1 (IRE1) [54, 59]. Under physiological conditions, the three transmembrane signal sensors bind to GRP78 on the membrane of the ER. During the URS, the misfolded proteins compete with the signal sensors to bind to GRP78, which triggers the signaling pathway of these three molecules [60]. Active PERK in turn activates the downstream signals, causing the decrease of protein translation and intracellular protein synthesis [55, 61]. ATF6 is separated from GRP78 and degraded in the Golgi apparatus to produce active fragments. The active fragments enter the nucleus to promote expression of UPR target genes and then modify or regulate protein folding [62, 63]. Active IRE-1 cleavages the X-Box Binding Protein 1 (XBP-1) mRNA gene precursor that activates ERAD and regulates protein translation [64]. In the early stage of ER stress, the activation of these three signaling pathways can protect the ER. However, continuous stress can aggravate oxidative stress and cell Ca 2+ overload, which cause cell apoptosis and excessive autophagy [54, 65].
Active ATF6, IRE1 and PERK promote the expression of the C/EBP homologous protein (CHOP). CHOP activates ER oxidoreductase 1(ERO1) to produce ROS [66]. c-Jun N-terminal kinase (JNK) is activated through the IRE1 pathway of the UPR and interacts with mitochondrial JNK-binding proteins, causing mitochondrial metabolism dysfunction and increasing level of ROS in mitochondria [67]. In addition, UPR leads to Ca2+ leakage from the ER and decrease of the Ca 2+ absorption, which in turn leads to Ca 2+ overload [68, 69]. The mitochondria reuptakes the ROS and excess Ca2+ produced by the ER, however this leads to the production of more ROS in the mitochondria. ER stress aggravates Ca2+ overload and oxidative stress, which eventually leads to apoptosis [70, 71]. Moreover, when UPR is activated, IRE1 activates the downstream nuclear transcription factor-κB (NF-κB), JNK and Recombinant X-Box Binding Protein 1 (XBP1) and cleaved ATF6, leading to the transcriptional activation of inflammation-related proteins; CHOP will also promote the release of proinflammatory cytokines. These factors contribute to the inflammatory reaction [72, 73].
ER stress is mediated by oxidative stress, inflammation and Ca 2+ overload, and ER stress will aggravate these pathological processes in turn. NPPs reduce MIRI by regulating ER stress-mediated signaling pathways,for example, Barbaloin pretreatment reduces MIRI by regulating the CNPY2-PERK apoptotic pathway [74]. Tournefolic acid B reduces MIRI by inhibiting PI3/Akt-mediated ER stress [75].
2.5. Inflammatory Response
During ischemia, inflammation is activated and further activated during reperfusion. Inflammation is an important cause of MIRI as it induces myocardial cell damage and affects myocardial function [76, 77].
During myocardial ischemia, a lot of cell debris and inflammatory factors are produced. Cell debris and inflammatory factors are known as damage associated molecular patterns (DAMPs). Some DAMPs, such as ATP, single-stranded RNA, mitochondrial DNA, double stranded RNA, and complement factors, are recognized and bound to pattern recognition receptors (PRRs) to trigger inflammatory responses [78, 79].
NLRP3 protein plays an important role in inflammasome activation. NLRP3 inflammasome, which is composed of NLRP3 receptor, apoptosis-associated speck-like protein containing a card domain (ASC) and Caspase-1, is a form of PRR and plays a vital role in inflammation caused by MIRI [80, 81]. NLRP3 expression is low under physiological conditions. During I/R, TLR ligands in cells are activated by DAMPs, which activate the NF-κB pathway through myeloid differentiation factor 88 (MyD88)-dependent or My88-independent pathways, and the activated NF-κB pathway in turn promotes the expression of NLRP3 [82-84]. DAMPs in cells can stimulate the assembly of the NLRP3 inflammasome and activate Caspase-1. Activated Caspase-1 promotes the cleavage of pro-IL-1β and pro-IL-18 into IL-1β and IL-18 respectively. IL-1β and IL-18 initiate an inflammatory cascade, which eventually leads to pyroptosis [85]. During reperfusion, myocardial cells produce ROS, which can activate the complement system and induce neutrophils accumulation in the infarct area. Cell adhesion molecules guide neutrophil migration to myocardial cells, leading to microcirculation obstruction. This promotes the production of proteolytic enzymes and ROS, thereby aggravating inflammation [86, 87]. In addition, ROS also induces neutrophils to produce a large amount of TNF-α, which activates the NF-κB pathway and promotes the release of inflammatory factors, such as IL-1, IL-6 and TNF-α, causing tissue damage [88]. Mitochondria plays a significant role in inflammatory response. Studies have shown that mitochondrial dysfunction and DNA leakage from broken mitochondria will activate the NLRP3 inflammasome and aggravate MIRI [89, 90]. Moreover, Ca 2+ overload is also an important factor in increasing the inflammation [91].
The inflammatory response is the result of the interaction of multiple factors and multiple pathways. At present, inhibiting the inflammatory response can be induced directly by inhibiting inflammation-related pathways. For instance, Cinnamic acid inhibits activation of the NLRP3 inflammasome and reduces MIRI [92]. In addition, Artemisinin can alleviate MIRI by inhibiting the production of ROS and the activity of the NLRP3 inflammasome [93].
2.6. Apoptosis
Apoptosis is a regulated form of programmed cell death; it is characterized by cell shrinkage, condensation of cytoplasm, nuclear shrinkage, and formation of apoptotic bodies [94]. Apoptosis is an important pathological mechanism involved in the entire MIRI process; it aggravates myocardial injury and leads to myocardial dysfunction [95].
Apoptotic pathways mainly include death receptor apoptosis pathway, mitochondrial apoptosis pathway, and ER pathway [96].
The death receptor apoptosis pathway refers to the process in which death receptors bind to related ligands on the cell surface, activates the caspase pathway, and triggers apoptosis. Death ligands are members of tumor necrosis factor (TNF) family, and receptors are members of tumor necrosis factor receptor (TNFR) family [97]. When the ligand binds to the receptor, it will activate caspase-8, which in turn activates downstream caspases, eventually causing cell apoptosis [96-98]. In the process of MIRI, the levels of TNF-α and TRAIL are increased, resulting in apoptosis.
Mitochondria plays a vital role in the process of apoptosis. After the apoptosis signal is activated, the permeability of the mitochondrial membrane will change, resulting in the leakage of Cyt c from the mitochondria into the cytoplasm. Cyt c activates Caspase-9, and then cleaves caspase-3, and finally triggers cell apoptosis [99]. The Bcl-2 protein family regulates apoptosis by controlling mitochondrial permeability. The anti-apoptotic proteins Bcl-2 reside in the mitochondrial outer membrane and inhibit the release of Cyt c, while the pro-apoptotic Bcl-2 proteins, Bax, can reside in the cytoplasm and translocate to the mitochondria after signal transduction, where it promotes release of Cyt c [100]. In addition, the loss of MMP and the burst of ROS will also cause the loss of Cyt c from mitochondria into the cytoplasm, triggering apoptosis [101].
ER stress is an important cause of apoptosis [54]. The excessive ER stress caused by I/R can increase the expression of CHOP protein, which in turn inhibits the expression of Bcl-2. ER stress also promotes the activation of Caspase-12 protein, eventually leading to apoptosis [102, 103]. In addition, Ca 2+ overload and oxidative stress caused by ER stress also leads to apoptosis.
Apoptosis is the result of the interaction of multiple factors in the process of MIRI, and thus inhibiting cardiomyocyte apoptosis is also an important way to alleviate MIRI. At present, many NPPs can alleviate MIRI by inhibiting cardiomyocyte apoptosis. Dihydroquercetin could inhibit apoptosis via the PI3K/Akt pathway to reduce MIRI [104]. Resina draconis inhibits apoptosis via regulating the miR-423-3p/ERK signaling pathway [105].
2.7. Autophagy
Autophagy is a cellular process in which cells remove and recycle misfolded or damaged proteins and organelles through lysosomes [106]. It is associated with both cell survival and death, and plays an important role in maintaining the normal structure and function of the heart [107].
During myocardial ischemia, dysfunctional mitochondrial energy metabolism will cause the accumulation of large amounts of hydrogen ions and fatty acid through increased glycolysis, which will eventually lead to the inhibition of glycolysis process and fatty acid utilization. At this time, autophagy can promote the generation of ATP by fatty acids and amino acids in the cytoplasm through the tricarboxylic acid cycle [107, 108]. Moreover, autophagy decreases apoptosis and protects the mitochondria membrane and cell membrane integrity [107, 109].
However, during reperfusion, autophagy may have negative effects on cardiomyocytes, such as causing cardiomyocyte apoptosis. Excessive autophagy may affect cardiomyocyte energy metabolism and protein synthesis [108, 110].
mTOR and Beclin-1 are the critical proteins in the autophagy process of MIRI. mTOR is mainly involved in the ischemic stage, and Beclin-1 is mainly involved in the reperfusion stage [107, 111]. During ischemia, reduced ATP synthesis activates AMPK, which in turn inhibits the activation of mTOR through the AMPK-mTOR pathway [112]. In addition, autophagy can also be promoted through AKT-mTOR pathways [112]. During reperfusion, Beclin1 becomes the major mediator of autophagy. Although the upregulation of Beclin1 is responsible for autophagy activation during reperfusion, the mechanism by which cardiac I/R injury activates Beclin1 is still being studied [113]. One possible mechanism is its association with the Bcl-2 protein, which modulates Beclin1-mediated autophagic response to nutrient deprivation in cardiac cells [114]. ROS are also inducers of Beclin1 and mediates autophagy during reperfusion. Increased ROS generation is a major promoter of autophagy during this phase. Antioxidant interventions significantly suppress Beclin1 up-regulation, suggesting that ROS play a vital role in mediating Beclin1 upregulation [115, 116]. During I/R, ER stress can lead to continuous UPR process and decreased Bcl-2 expression, and eventually lead to excessive autophagy [117].
Autophagy is a double-edged sword in the process of MIRI. NPPs can reduce MIRI by promoting or inhibiting autophagy. For example, Gastrodin can increase autophagy by regulating the AMPK-mTOR pathway to reduce MIRI [118], and Ginsenoside Rb1 can inhibit autophagy by regulating the PI3K/Akt/mTOR pathway to alleviate MIRI [119].
3. Natural Plant Products in the Treatment of MIRI
3.1. Polyphenols
Polyphenols are a group of chemical substance; they contain multiple phenolic hydroxyl groups nad include more than 10,000 chemical compounds in a diverse variety of plants [120]. Polyphenols can be classified into different groups based on the number of phenol rings and the structural elements that bind these rings. They are classified into several subclasses, such as the lignans, stilbenes, flavonoids and phenolic acids [121, 122]. In recent years, numerous biological activities and healthy benefits of polyphenols have been reported, including anticancer activity, antioxidant activity, heart protection effect and anti-inflammatory activity [120, 123, 124]. Besides, many studies showed that polyphenols are important in alleviating MIRI by regulating oxidative stress, inflammation and other mechanisms, as shown in Table 1.
Table 1. The cardioprotective effects of polyphenols in MIRI.

Table 1. (Continued) The cardioprotective effects of polyphenols in MIRI.
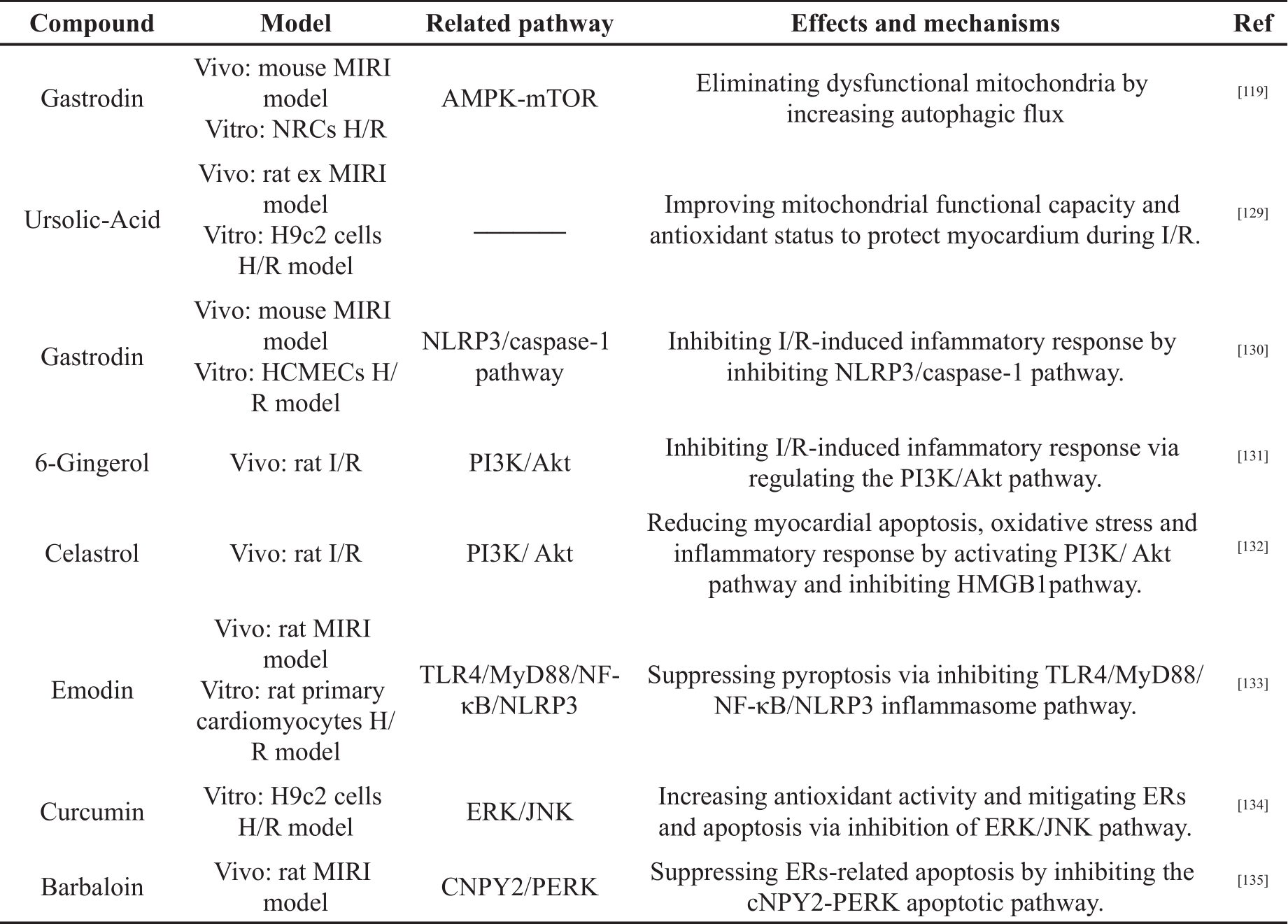
Chen et al. [125] ligated the left anterior descending artery (LAD) in male Wistar rat to construct the I/R model. Carvacrol (CAR) was injected after ligation. The H/R model was established in vitro using neonatal rat cardiomyocytes (NRCs). The results showed that CAR administration had significant effects against oxidative stress and apoptosis. The results showed that CAR up-regulated the phosphorylation of extracellular-regulated protein kinases (p-ERK) and down-regulated the phosphorylation of p38 and JNK. The cardioprotective effect of CAR was reversed by the ERK inhibitor, indicating that the MAPK/ERK pathway was involved in the anti-apoptotic mechanism of CAR. In addition, pretreatment with CAR activated the Akt/endothelial nitric oxide synthase (eNOS) pathway in H/R cardiomyocytes in vitro, but the protective effect of CAR was abolished by the addition of an Akt inhibitor. These results suggested that the cardioprotective effect of CAR may be attributed to its antioxidant and anti-apoptotic activities through the regulation of MAPK/ERK and Akt/eNOS signaling pathways.
Tian et al. [126] treated H9c2 cardiomyocytes with H 2O 2 to establish an oxidative damage model. Pretreatment with calendula E (CE) analogue suppressed production of ROS, maintained MMP and inhibited cardiomyocyte apoptosis. Furthermore, CE analogues could increase the expression of B-cell lymphoma-2 (Bcl-2) and decrease the expression of Bcl-2-associated x protein (Bax) in a dose-dependent manner. These results suggested that CE analogues could protect H9c2 cardiomyocytes from H 2O 2-induced oxidative stress and apoptosis by inhibiting ROS production in cardiomyocytes and maintaining the stability of mitochondrial energy metabolism.
Yang et al. [127] established ex vivo I/R model and in vitro H/R models. The results showed that Vitexin (VT) inhibited production of ROS and the expression of exchange protein directly activated by cyclic adenosine monophosphate (cAMP) (Epac1) and Drp1. It also increased Mfn2 expression and MMP, thereby inhibiting myocardial cell apoptosis. These results suggested that VT inhibits cardiomyocyte apoptosis by regulating the Epac1 pathway to inhibit mitochondrial dysfunction.
Wang et al. [128] investigated the antioxidant effect and the underlying pathways of Platycodin D (PD) during I/R in H9c2 cells H/R model. The results showed that PD reduced production of ROS and malondialdehyde (MDA), and increased SOD production. Pretreatment with PD reduced the expression of Bax, cleaved caspase-3 and improved Bcl-2 expression. Furthermore, PD was found to induce the activation of Akt/nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase 1(HO-1) pathway, while the inhibitor of Akt reduced the effects of PD on the H/R model. These results indicated that PD pretreatment could inhibit H/R-induced cell apoptosis and oxidative stress by regulating the Akt/Nrf2/HO-1 pathway.
Wu et al. [52] constructed MIRI models of C57 mice and ex MIRI models of rat in vivo. H/R models of H9c2 cells and NRCs were constructed in vitro. After pretreatment with Icariin, it was found that Icariin could alleviate MI; inhibit cardiomyocyte apoptosis; restore cardiac function; reduce the level of lactate dehydrogenase (LDH), MDA and creatine kinase-MB (CK-MB); increase SOD content; and increase the expression of silent information regulator 1 (SIRT-1) and Bcl-2. Moreover, the expression of cleaved-caspase-3, acetyl-recombinant forkhead Box Protein O1 (Ac-FOXO1) and Bax protein decreased. However, the effect of Icariin was abolished by inhibitors of SIRT-1. These results suggested that Icariin protected against I/R-induced oxidative stress injury by regulating the SIRT-1/FOXO1 signaling pathway.
Shi et al. [51] used the Langendorff method to construct MI/R model and injected hematoxylin A (SA) before reperfusion. The results showed that SA postconditioning reduced myocardial infarct size, and inhibited LDH, CK-MB and cardiac troponin (cTnI) release. Other findings were that SA enhanced myocardial ATP production, enhanced mitochondrial complex activity, prevented loss of MMP and prevented opening of MPTP. In addition, SA treatment increased mitochondrial DNA (mtDNA) copy number and expression of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), Mfn2 and LC3-Ⅱ, however, it decreased the expression of Drp1. Adenosine monophosphate activated protein kinase (AMPK) antagonist could eliminate the protective effect of SA on the isolated heart. These results suggested that SA could reduce I/R induced-mitochondrial dysfunction and MI by regulation of mitochondrial quality control through activation of the AMPK signal pathway.
Fu et al. [117] constructed I/R model of C57 mice in vivo and NRCs H/R model in vitro to investigate the protective effect and potential mechanism of Gastrodin (GAS) during MIRI. The results showed that GAS could inhibit cardiomyocyte apoptosis. Expression of LC3Ⅱ and p-MAPK were increased, while expression of P62 and phosphorylation of the mechanistic target of rapamycin (P-mTOR) were reduced. The MMP was maintained. These results suggested that GAS reduced mitochondrial damage through autophagy and reduced I/R-induced apoptosis and tissue damage.
Yu et al. [129] explored the potential mechanisms by which ursolic-acid (UA) reduced MIRI in the H9c2 cells H/R model and rat ex MIRI model. The results showed that UA could enhance mitochondrial ATP synthesis and glutathione redox cycling to inhibit myocardial apoptosis. These results indicated that UA could protect the myocardium during I/R by improving mitochondrial functional capacity and antioxidant status.
Zhu et al. [27] explored the mechanism of Luteolin (Lut) in reducing Ca 2+ overload during I/R in a rat ex MIRI model. The results showed that Lut increased the expression of phospholamban (p-PLB) and SERCA, and decreased the expression of p-MAPK. In addition, Luteolin could restore MMP and inhibit apoptosis. These results suggested that Lut could enhance SERCA activity by activating p-PLB via inhibition of the MAPK pathway; ER reabsorption of Ca 2+ improves, and mitochondrial damage caused by Ca 2+ overload and apoptosis are inhibited.
Ye et al. [39] discovered HSYA could inhibit the sharp increase of the Ca 2+ current induced by H/R, maintain the stability of intracellular Ca 2+ concentration and reduce mitochondrial membrane potential by regulating L-type calcium channel (LTCC). HSYA could inhibit the expression of Bax and caspase-3 protein, and promote the expression of Bcl-2 protein. These results demonstrated that HYSA can alleviate MIRI through its inhibitory effect on Ca 2+ overload and myocardial cell apoptosis.
Sun et al. [104] established the I/R model in male mice and the H/R model in human cardiac microvascular endothelial cells (HCMECs). Gastrodin (100 mg/kg/day) was injected intraperitoneally for 3 days before modeling. GAS (20, 40, 80, 100 μM) was given to the cells 2 hours before reoxygenation. The results showed that GAS pretreatment could increase the expression of VEGF protein in vitro and in vivo, while inhibiting the expression of IL-1β, NLRP3 and caspase-1 protein. GAS can also inhibit cell apoptosis and pyroptosis, and reduce myocardial infarction size. These results indicated that GAS could reduce the inflammatory response by inhibiting the NLRP3/caspase-1 pathway and myocardial tissue cell apoptosis to reduce myocardial injury.
Xu et al. [131] used MIRI model in SD rat. 6-gingerol (6-G) (6 mg/kg) was injected into the tail vein 30 minutes before LAD ligation. In the other group, 6-G (6 mg/kg) and a PI3/AKT inhibitor (LY) were administered via the tail vein 30 minutes before LAD ligation. 6-G preconditioning could reduce the area of myocardial injury and maintain physiological cardiac contractility. The level of TNF-α, IL-1β, IL-6 and CK-MB in serum were reduced. It also inhibited the expression of NLRP3, TNF-α, IL-1β and IL-6 in myocardial tissue and increased the expression of P-AKT and PI3. However, in the 6-G+LY group, the experimental results above were reversed. These results indicated that 6-G can inhibit IR-induced inflammation and reduce myocardial tissue damage through the PI3/AKT pathway.
Tong et al. [132] explored the relationship between triptolide and inhibition of I/R-induced inflammation in rat MIRI model. The results showed that Celastrol preconditioning could reduce myocardial infarction size. It can inhibit the expression of high mobility group box 1 (HMGB1), LC3, Beclin-1, Bax and p-AKT; increase the expression of Bcl-2; and reduce the level of inflammatory factors. However, the protective effect of Celastrol on myocardial tissue was reduced after adding a P13k inhibitor. The results showed that Celastrol alleviates myocardial I/R injury through the PI3K/Akt pathway to inhibit HMGB1 expression, thereby reducing myocardial apoptosis, myocardial autophagy and the inflammatory response.
Ye et al. [133] constructed a rat MIRI model in vivo and H/R model in vitro to investigate the effects of Emodin on MIRI. The expression of NLRP3 and its downstream inflammatory factors were significantly up-regulated in H/R-treated cardiomyocytes, while emodin pretreatment inhibited the expression of these proteins. Emodin also inhibited cell pyroptosis, improved cell survival rate in vitro and reduced myocardial infarction size in rat. These results indicated that emodin could inhibit the inflammatory response induced by H/R and inhibit pyroptosis.
Wei et al. [134] investigated the effect of Curcumin (Cur) on MIRI in H9c2 H/R model. The results showed that Cur inhibited cardiomyocyte apoptosis; decreased the level of LDH and MDA; and increased the level of SOD. Besides, Cur decreased the mRNA expression of GRP78 and CHOP, and the expression of GRP78, CHOP, p-ERK1/2 and p-JNK. These results demonstrated that Cur could reduce MIRI by regulating ER-related pathways and inhibiting oxidative stress.
Cui et al. [74] investigated MIRI in a rat model and showed that BAR could reduce MI, and reduce the expression of ER stress-related proteins (GRP78, CHOP, caspase-3/12, p-PERK, ATF4). In addition, BAR inhibited the mRNA expression of protein canopy homolog 2 (CNPY2) and GRP78. These results suggested that BAR could alleviate MIRI by inhibiting the CNPY2/PERK apoptotic pathway.
Jo et al. found that the effect of curcumin on MIRI is inconsistent. The experimental results showed that compared with the model group, curcumin administration could not significantly reduce the myocardial infarction area, because LDH, CK-MB and AST activity are still high. Futher more, curcumin could not effectively improve cardiac function [135]. In a study on Bifunctional Supramolecular Hydrogel in the context of MIRI, curcumin did not effectively inhibit autophagy and apoptosis caused by MIRI. However, curcumin embedded in the hydrogel had a good effect on alleviating MIRI [136].
3.2. Saponins
Saponins are a structurally diverse group of bioactive NPPs, and they are polar molecules, containing triterpene or steroid aglycone with one or more sugar chains [137, 138]. Many Chinese herbal medicines contain large amounts of saponins, such as Panax ginseng C. A. Mey, Panax notoginseng (Burk.) F.H. Chen, Panax quinquefolium L, Aralia elata (Miq.), and Astragali Radix [139- 143]. In recent years, studies on saponins have found that they have antioxidant and cardiovascular protection properties, and can be used for treatment of neurological diseases [144, 145]. In addition, many studies have shown that saponins can alleviate MIRI, as illustrated in Table 2.
Table 2. The cardioprotective effects of saponins in MIRI.
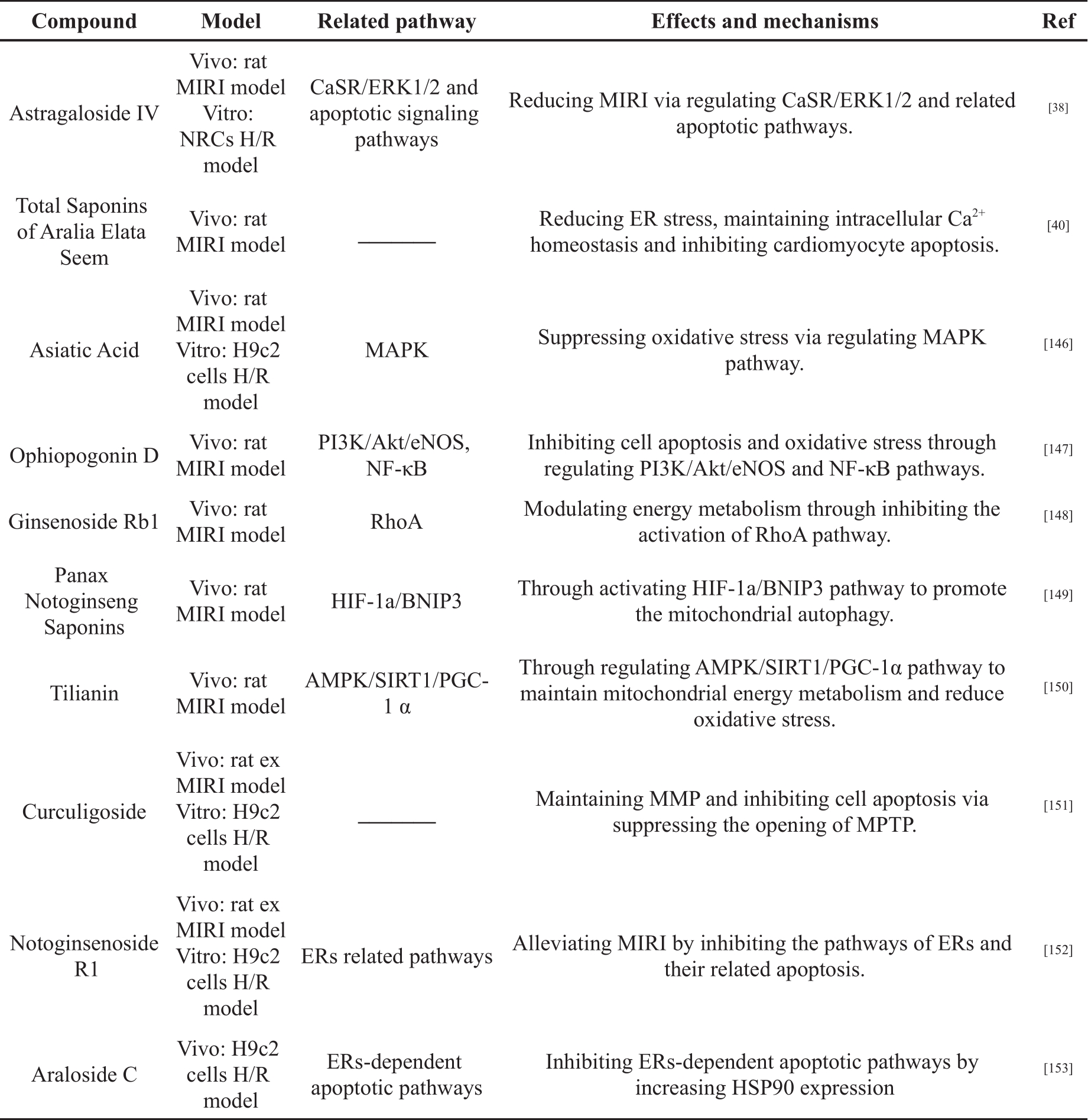
Yi et al. [146] studied the mechanism by which asiatic acid (AA) is able to reduce MIRI in rat MIRI model and H9c2 cell H/R model. The results showed that AA could reduce the contents of MDA and ROS in serum and cells, and increase the level of SOD. The mitochondrial morphology and membrane potential were maintained. Reduction of cell apoptosis was achieved by inhibiting p38 and JNK phosphorylation; increasing Bcl-2/Bax ratio; reducing Cyt c release; and inhibiting the caspase cascade. These results suggested that AA exerts cardioprotective effects through MAPK regulation of ROS-induced oxidative stress.
Huang et al. [147] constructed a rat MIRI model to investigate the mechanism of Ophiopogonin D (OP-D) in reducing MIRI. The results showed that OP-D could reduce myocardial injury, restore cardiac function and inhibit myocardial cell apoptosis in rat. OP-D treatment increased the expression of P-AKT and P-eNOS, and inhibited the expression of P-JNK. In addition, OP-D inhibited the expression of NF-κB in the nucleus. mRNA expression of Epoxyeicosatrienoic acids (EETs) and cytochrome P450 epoxygenase (CYPs) were increased. These results suggested that OP-D could alleviate cardiac injury after MI/R injury by regulating PI3K/Akt/eNOS and NF-κB signaling pathways to inhibit cell apoptosis and oxidative stress.
Cui et al. [148] investigated the effect of Ginsenoside Rb1 (Rb1) on rat MIRI model. The results showed that Rb1 treatment inhibited myocardial structural disruption, cardiomyocyte apoptosis, myocardial infarction and myocardial cTnI release induced by I/R. Rb1 also inhibited activation of the Ras homolog gene family member A (RhoA) signaling pathway and restored ATP production during myocardial I/R. In addition, surface plasmon resonance (SPR) experiments showed that Rb1 inhibited RhoA in a dose-dependent manner. The results suggested that Rb1 could restore mitochondrial energy metabolism and attenuate MIRI by regulating the RhoA signaling pathway.
Liu et al. [149] explored the relationship between Panax Notoginseng Saponins (PNS) and mitochondria by constructing a rat MIRI model. The results showed that PNS was able to reduce CK, MDA and LDH in serum and increase SOD in myocardial tissue. PNS treatment also increased the expression of hypoxia inducible factor (HIF-1α), Bcl2 interacting protein 3 (BNIP3), Beclin-1 and autophagy protein 5 (Atg5), as well as increasing the ratio of LC3 Ⅲ/LC3Ⅰ. In addition, PNS treatment improved mitochondrial morphology and inhibited cardiomyocyte apoptosis. The results indicated that PNS could alleviate MIRI by appropriately increasing the autophagic capacity of mitochondria and reducing ROS production.
Tian et al. [150] explored whether Tilianin was able to alleviate MIRI by regulating mitochondrial energy metabolism and oxidative stress through the AMPK/SIRT1/peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) signaling pathway in the rat MIRI model. The results showed that Tilianin could reduce myocardial infarct size and improve myocardial pathological morphology. It could increase levels of ATP and NAD +; enhance SOD activity; and significantly increase AMPK, SIRT1 and PGC-1α mRNA levels. Meanwhile, it could decrease AMP/ATP ratio, MDA and ROS. The protein expression of AMPK, p-AMPK, SIRT1, PGC-1α, recombinant nuclear respiratory factor 1 (NRF1), recombinant transcription factor A, Mitochondrial (TFAM) and FOXO1 were up-regulated. However, these effects were inhibited by AMPK-specific and SIRT1-specific inhibitors, respectively; this suggests Tiliani alleviates MIRI through regulation of the AMPK/SIRT1/PGC-1α signaling pathway to maintain mitochondrial energy metabolism and reduce oxidative stress.
Zhao et al. [151] constructed rat ex MIRI model in vivo and H9c2 H/R model in vitro to investigate the effect of Curculigoside on MIRI. The results showed that pretreatment with Curculigoside significantly increased cell viability, inhibited cardiomyocyte apoptosis and reduced infarct size. Curculigoside also inhibited the opening of MPTP and maintained ΔΨm. In addition, curculigoside significantly reduced the expression of apoptosis-related proteins. Notably, the protective effect of curculigoside was abolished by the MPTP opener. These results suggested that curculigoside may prevent MIRI by inhibition of mitochondrial MPTP opening, MMP maintenance and cell apoptosis inhibition.
Yin et al. [38] found that AsIV pretreatment in NRCs H/R model could significantly improve cell viability and cardiomyocyte survival. Reductions were seen in lactate dehydrogenase (LDH) release, intracellular free calcium concentration ([Ca 2+]i) and expression of CaSR, while phosphorylation of ERK1/2 increased. Comparatively, in a rat model of MI/R, pretreatment with AsIV significantly reduced the area of cardiac injury and cardiomyocyte apoptosis. Reductions were seen in CK-MB and cTnI levels. These results suggested that AsIV alleviates MIRI by inhibiting the CaSR/ERK1/2-related apoptotic signaling pathway.
Wang et al. [40] used a rat MIRI model to investigate the effects and mechanism of saponins, found in Aralia elata (Miq) seem (AS), in the context of MIRI. The results showed that the AS could significantly reduce the myocardial infarction area, and the levels of LDH, CK and MDA. It increased the content of SOD, and restored Ca 2+-Mg 2+-ATPase, Na +-K +-ATPase, Serca2a and calcineurin (CaN). The expression of Bcl-2 was up-regulated and the expression of Bax was down-regulated in the same treatment group. These results indicated that AS could reduce ER stress, maintain intracellular Ca 2+ homeostasis, inhibit cardiomyocyte apoptosis and alleviate MIRI injury.
Yu et al. [152] investigated the cardioprotective effect of Notoginsenoside R1 (NGR1) on rat ex MIRI model and H9c2 cell H/R model. The results showed that NGR1 pretreatment could reduce myocardial injury, restore cardiac function and inhibit cell apoptosis. NGR1 decreased the expression of ER stress responsive proteins, including GRP78, P-PERK, ATF6 and IRE, and inhibited the expression of proapoptosis proteins, including CHOP, Caspase-12 and P-JNK. Moreover, NGR1 reduced ROS level and increased the activity of antioxidants. These results indicated that NGR1 could alleviate MIRI by inhibiting the ER stress-related pathways.
Du et al. [153] investigated the effect of Araloside C (AsC) on H/R-induced apoptosis in H9c2 cells H/R model. These results showed that pretreatment with AsC could inhibit the leakage of LDH and cell apoptosis. AsC also inhibited the activation of the PERK/eIF2α pathway and decreased the expression of CHOP and caspase-12. Additionally, AsC significantly increased the expression of HSP90. The suppression effect of AsC on ER stress-related apoptosis, caused by H/R, is blocked by an HSP90 inhibitor. In conclusion, AsC can reduce H/R-induced apoptosis through increasing HSP90 expression to inhibit ER stress-dependent apoptotic pathways.
3.3. Terpenoids
Terpenoids, also known as terpenes or isoprenoids, are the general term for compounds and their derivatives with an isoprene unit as the basic structural unit [154, 155]. At present, there are more than 80,000 terpenoids, which are mainly extracted from plants and bacteria [154, 156]. Many Chinese herbal medicines contain large amounts of terpenoids, such as Euphorbia lathyrism L, Rhizoma Alismatis and Salvia miltiorrh [157- 159]. Terpenoids are anti-inflammatory, anti-oxidative stress, and regulate mitochondrial function [160, 161]. In addition, there have been many advances in the use of terpenoids in alleviating MIRI, as shown in Table 3.
Table 3. The cardioprotective effects of terpenoids in MIRI.

Chen et al. [162] established an H/R model in H9c2 cardiomyocytes. Pretreatment with Tanshinone IIA (TSA) show inhibition of apoptosis of cardiomyocytes; reduction in the level of LDH, MDA and ROS; and increased MMP. Knockdown of AK003290 and overexpression of miR-124-5p reversed the anti-H/R effect of TSA. These results showed that TSA could improve MMP, inhibit oxidative stress, reduce cardiomyocyte apoptosis, and alleviated H9c2 cardiomyocytes injury indued by H/R, by regulating the lncRNA AK003290/miR-124-5p pathway.
Zhuo et al. [163] used tert-butyl hydroperoxide (t-BHP)-stimulated H9c2 cell model and rat MIRI model to investigate the cardioprotective effect of Tanshinone I (TI). The results showed that TI could inhibit the phosphorylation of receptor interacting protein kinase 1 (p-RIP1), phosphorylation of the receptor interacting protein kinase 3 (p-RIP3) and phosphorylation of mixed lineage kinase domain-like (p-MLKL). Meanwhile, TI can increase the expression of Nrf2, HO-1 and other antioxidant related proteins; reduce ROS production; and reverse MMP loss in the in vitro model. In the in vivo MIRI model, TI could alleviate the degree of myocardial tissue injury; reduce the level of TNF-α, MDA and IL-6; and increase the level of SOD. These results suggested that TI could inhibit oxidative stress and cellular inflammation by suppressing RIP1/RIP3/MLKL and activating Akt/Nrf2 pathways to protect the heart.
3.4. Alkaloids
Alkaloids are a class of secondary products that are widely present in plants. More than 12,000 different structures have been identified, most of which contain nitrogen atoms [167, 168]. Alkaloids have a wide range of pharmacological activities and many of them have been applied in traditional or modern medicine [164]. In recent years, studies on alkaloids have found that they are anti-inflammatory, anti-cancer and anti-arrhythmic [165- 167]. In addition, many studies have also proven that alkaloids have a significant effect on alleviating MIRI, and the main mechanism of this is through inhibiting myocardial cell apoptosis and improving mitochondrial energy metabolism, as shown in Table 4.
Table 4. The cardioprotective effects of alkaloids in MIRI.

Zhu et al. [168] explored the effect of Berberine (BBR) on MIRI by constructing an H/R model using H9c2 cells and rat MIRI model. In vitro, BBR pretreatment could regulate the expression of autophagy-related proteins, induce the production of autophagosomes and suppress the increase of MMP. In vivo, BBR reduced myocardial infarct size, inhibited cardiomyocyte apoptosis, and significantly reduced the level of CK-MB, LDH, and AST. In addition, the effect of BBR on MIRI was weakened after BNIP3 knockdown. Luciferase reporter and ChIP assays showed that BBR regulated BNIP3 expression through enhancing the binding of HIF-1α to the BNIP3 promoter. These results suggested that BBR moderately enhanced autophagy and inhibited cardiomyocyte apoptosis by regulating the HIF-1α/BNIP3 pathway, thus alleviating MIRI.
Zhao et al. [169] investigated the effect of BBR on MIRI by constructing a rat MIRI model and an H9c2 H/R model. The results showed that BBR pretreatment significantly reduced myocardial infarction size; maintained cardiac function; inhibited myocardial apoptosis; down-regulated phosphorylation of PERK and 65kDa eukaryotic translation initiation factor 2A (eIF2α); and down-regulated the expression of ATF4 and CHOP in cardiac tissues and H9c2 cells. In addition, pretreatment with BBR increased the phosphorylation of JAK2 and human signal transducer and activator of transcription 3 (STAT3). The inhibitory effect of BBR on ER stress and its cardiac protection effect could be abolished by JAK2/STAT3 specific inhibitor or transfection of JAK2 siRNA. These results suggested that BBR inhibited ER stress by activating JAK/STAT3 pathway to reduce MIRI.
4. Clinical Evidence
In view of the great potential of NPPs in reducing MIRI, clinical trials have been carried out to further verify whether certain NPPs can reduce myocardial injury following elective percutaneous coronary intervention.
Wongcharoen et al. [170] randomly divided 121 patients into two groups; one group received placebo and the other group received curcuminoids 4 g/day, in addition to standard therapy, from 3 days before coronary artery bypass grafting (CABG) to 5 days after CABG. The results showed that curcumin could significantly reduce CPR and MDA in serum, and reduce the incidence of left ventricular dysfunction and NT-proBNP. This proved that curcumin has the ability to reduce MIRI clinically. However, the study by Aslanabadi et al. [170] failed to support the ability of curcumin to alleviate MIRI. Aslanabadi et al. divided 110 patients into standard treatment group and intervention group. In addition to standard treatment, the intervention group were given oral administration of 480 mg nanomicelle curcumin before PCI. The results showed that the intervention group did not have significant reductions in serum levels of CK-MB and cTn-I. Asgary et al. [171]. found that treatment with lycopene (30 mg 12 hours before PCI, 15 mg before PCI and 8 hours after PCI) added to the standard treatment group significantly reduced serum level of CK-MB, but did not significantly reduce level of cTn-I, compared to standard treatment. The study suggests that lycopene may alleviate MIRI. Aslanabadi et al. [172] randomly divided 99 patients into intervention group and control group. In the intervention group, 300 000 IU of vitamin D was taken orally 12 hours before PCI. The experimental results showed that vitamin D could not effectively reduce the serum levels of CK-MB and cTn-I when compared with the control group.
5. Discussion
5.1. Contradictions between Different Experiments
This review included 35 preclinical studies and 4 clinical studies. Thirty-three preclinical studies have shown that NPPs have the potential to alleviate MIRI, and two of them have inconsistent views. This may be related to the poor water solubility of the NPPs, especially curcumin. Embedding curcumin in the hydrogel increased the solubility of curcumin in water, and significantly improved the effectiveness of curcumin on MIRI [136].
Among the clinical research, we found that there were differences in experimental results. The studies of Wongcharoen et al. [170] and Asgary et al. [172] supported the role of NPPs in clinically reducing MIRI, however, Aslanabadi et al [171] and Aslanabadi et al. [173] could not support the effectiveness of NPPs in MIRI. It should be noted that the studies with positive experiemtnal evidence used larger doses of NPPs and had longer treatment periods. If the administration period of NPPs is short and the dose is low, it’s difficult for NPPs to exert protective effects on the heart following PCI. Due to the lack of research on the active ingredients of NPPs, and the shortcomings in bioavailability and affinity of the active ingredients, the therapeutic effect of NPPs administration is slow [174]. Changing the dosage form of drugs may be necessary to achieve and maintain an effective blood concentration for therapeutic purposes [175].
At present, there is no good method to alleviate MIRI. Most of the studies mentioned in this article have involved pre-administration of the NPP for a long time before ischemia, due to the design of animal or cell experiments. Thus, NPPs can play a preventive role in MIRI and this has been proved in clinical practice. For example, curcumin can be administered before PCI to protect the heart, reduce the incidence of myocardial infarction, and reduce the level of biomarkers after PCI [170].
5.2. Limitation
The preclinical studies of NPPs to alleviate MIRI lack monitoring of cardiac function, so the experimental results cannot directly reflect the protective effect of NPPs on the heart. It may be necessary to conduct drug administration in animals after reperfusion and monitor the cardiac function of animals for a longer period of time in future preclinical studies.
At present, there are few clinical studies on the use of NPPs to alleviate MIRI. The research protocols of the above clinical studies are relatively simple, and there are few studies involving cardiac function tests. In the future, more clinical studies are needed to monitor the incidence of myocardial infarction and cardiac function for a longer time.
Experimental data is collected shortly after ischemia and reperfusion in most animal experiments and cell experiments, which means most experiments do not administrate long-term drug treatment after reperfusion. In summary, preclinical studies suggested that pretreatment with NPPs could protect the heart from I/R induced myocardial injury. However, there are some differences between the drug delivery methods in the studies and clinical drug delivery requirements; it is necessary to further explore drug delivery methods in animal models, in order to establish appropriate clinical drug delivery methods.
6. Conclusion
NPPs are multi-target compounds and they have shown great therapeutic potential. In recent years, many studies have been conducted on the use of NPPs to reduce MIRI. The composition of NPPs is complex, and its anti-MIRI mechanisms are diverse. This review is mainly based on the material structure of NPPs and their mechanisms of alleviating MIRI. Some components of NPPs have anti-inflammatory and anti-oxidative properties, thus they can inhibit I/R-induced oxidative stress, Ca 2+ overload, inflammation, mitochondrial dysfunction and ER stress. MIRI is the result of the interaction of multiple factors and multiple signaling pathways. The complex signaling molecular regulation of MIRI requires drugs that act on multiple targets and pathways. NPPs act on multiple pathways at the same time to effectively reduce MIRI, as shown in Figure 1. Therefore, it is highly possible for NPPs to be developed into potent anti-MIRI drugs, with low toxicity. At present, the research on reducing MIRI using NPPs is mainly focused on flavonoids and saponins. The relationship between the chemical structure of NPPs and its anti-MIRI bioactivity remains to be discovered. Understanding of the molecular mechanisms underlying the activities of different types of NPPs against MIRI has gradually deepened; however, most of the studies are limited to animal and cell models, and research into the clinical application of NPPs is limited.

Figure 1. NPPs can inhibit MIRI-induced mitochondrial dysfunction, Ca 2+ overload, ER stress, oxidative stress and inflammatory reaction by regulating signal pathways.
There are some shortcomings in the safety of NPPs in clinical application, including the following aspects: the mechanism of action of NPPs is complex; the active ingredients are difficult to determine; the dose and frequency of use are difficult to control; it may have poor efficacy or excessive reactions. In addition, the drug utilization of NPPs is generally low, thus larger doses and longer administration cycles are required, and this may cause toxic reactions [175]. There are many interactions and interference factors between NPPs and other therapeutic drugs, which need to be evaluated and controlled in detail, according to the physiological characteristics and conditions of specific patients.
Future research on the use of NPP for treatment of MIRI needs to further explore NPPs’ anti-MIRI mechanism and the structure-activity relationship of NPPs. It is necessary and urgent to translate new research into clinical treatment programs.
Author Contributions: investigation, Peixun Yang; writing—original draft preparation, Peixun Yang; writing—review and editing, Minxuan Liu and Xiaoxue Fan; supervision, Xinzhuang Zhang, Liang Cao and Zhenzhong Wang; project administration, Wei Xiao; All authors have read and agreed to the published version of the manuscript.
Funding: This work was supported by Programs Foundation for Leading Talents in National Administration of Traditional Chinese Medicine of China “Qihuang scholars” Project.
Data Availability: Statement: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.
References
- Nowbar A. N. ; Gitto M. ; Howard J. P. ; et al . Mortality from ischemic heart disease. Circ. Cardiovasc. Qual. Outcomes, 2019, 12( 6): e005375. DOI: https://doi.org/10.1161/CIRCOUTCOMES.118.005375
- Roth G. A. ; Mensah G. A. ; Johnson C. O. ; et al . Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J. Am. Coll. Cardiol., 2020, 76(25): 2982- 3021.
- Johnson T. ; Zhao L. ; Manuel G. ; et al . Approaches to therapeutic angiogenesis for ischemic heart disease. J. Mol. Med (Berl)., 2019, 97(2): 141- 151. DOI: https://doi.org/10.1007/s00109-018-1729-3
- Zhang X. L. ; Zhu Q. Q. ; Yang J. J. ; et al . Percutaneous intervention versus coronary artery bypass graft surgery in left main coronary artery stenosis: a systematic review and meta-analysis. BMC. Med., 2017, 15(1): 84. DOI: https://doi.org/10.1186/s12916-017-0853-1
- Prem P. N. ; Sivakumar B. ; Boovarahan S. R. ; et al . Recent advances in potential of Fisetin in the management of myocardial ischemia-reperfusion injury-A systematic review. Phytomedicine, 2022, 101: 154123. DOI: https://doi.org/10.1016/j.phymed.2022.154123
- He J. ; Liu D. ; Zhao L. ; et al . Myocardial ischemia/reperfusion injury: Mechanisms of injury and implications for management (Review). Exp. Ther. Med., 2022, 23(6): 430.
- Wang R. ; Wang M. ; Zhou J. ; et al . Saponins in Chinese herbal medicine exerts protection in myocardial ischemia-reperfusion injury: possible mechanism and target analysis. Front. Pharmacol., 2020, 11: 570867. DOI: https://doi.org/10.3389/fphar.2020.570867
- Wang R. ; Wang M. ; He S. ; et al . Targeting calcium homeostasis in myocardial ischemia/reperfusion injury: an overview of regulatory mechanisms and therapeutic reagents. Front. Pharmacol., 2020, 11: 872. DOI: https://doi.org/10.3389/fphar.2020.00872
- Su X. ; Zhou M. ; Li Y. ; et al . Mitochondrial damage in myocardial ischemia/reperfusion injury and application of natural plant products. Oxid. Med. Cell. Longev., 2022, 2022: 8726564.
- Zhao C. ; Li S. ; Zhang J. ; et al . Current state and future perspective of cardiovascular medicines derived from natural products. Pharmacol. Ther., 2020, 216: 107698. DOI: https://doi.org/10.1016/j.pharmthera.2020.107698
- Bu W. ; Zhang Z. ; Ocansey D . K. W.; et al. Research on natural products from traditional Chinese medicine in the treatment of myocardial ischemia-reperfusion injury. Am. J. Transl. Res., 2022, 14(3): 1952-1968.
- Cadenas S . ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free. Radic. Biol. Med., 2018, 117: 76-89. DOI: https://doi.org/10.1016/j.freeradbiomed.2018.01.024
- Kurian G. A. ; Rajagopal R. ; Vedantham S. ; et al . The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid. Med. Cell. Longev., 2016, 2016: 1656450. DOI: https://doi.org/10.1155/2016/1656450
- Navarro Yepes. J. ; Burns M. ; Anandhan A. ; et al . Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid. Redox. Signal., 2014, 21(1): 66-85. DOI: https://doi.org/10.1089/ars.2014.5837
- Gopalakrishnan B. ; Nash K. M. ; Velayutham M. ; et al . Detection of nitric oxide and superoxide radical anion by electron paramagnetic resonance spectroscopy from cells using spin traps. J. Vis. Exp., 2012(66): e2810. DOI: https://doi.org/10.3791/2810
- Andreadou I. ; Schulz R. ; Papapetropoulos A. ; et al . The role of mitochondrial reactive oxygen species, NO and H 2S in ischaemia/reperfusion injury and cardioprotection . J. Cell. Mol. Med., 2020, 24(12): 6510-6522. DOI: https://doi.org/10.1111/jcmm.15279
- Peoples J. N. ; Saraf A. ; Ghazal N. ; et al . Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med., 2019, 51( 12): 1- 13. DOI: https://doi.org/10.1038/s12276-019-0355-7
- Neri M. ; Riezzo I. ; Pascale N. ; et al . Ischemia/reperfusion injury following acute Myocardial Infarction: A critical issue for clinicians and forensic pathologists. Mediators. Inflamm., 2017, 2017: 7018393. DOI: https://doi.org/10.1155/2017/7018393
- Ho E. ; Karimi Galougahi. K. ; Liu C. C. ; et al . Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox. Biol., 2013, 1: 483-91. DOI: https://doi.org/10.1016/j.redox.2013.07.006
- Halestrap A. P. ; Kerr P. M. ; Javadov S. ; et al . Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim. Biophys. Acta., 1998, 1366(1-2): 79-94. DOI: https://doi.org/10.1016/S0005-2728(98)00122-4
- Van . Der. Pol. A.; Van Gilst. W. H.; Voors A. A.;et al.; Treating oxidative stress in heart failure: past, present and future. Eur. J. Heart. Fail., 2019, 21(4): 425-435. DOI: https://doi.org/10.1002/ejhf.1320
- Anzell A. R. ; Maizy R. ; Przyklenk K. ; et al . Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol. Neurobiol., 2018, 55(3): 2547-2564. DOI: https://doi.org/10.1007/s12035-017-0503-9
- Yu D. ; Li M. ; Tian Y. ; et al . Luteolin inhibits ROS-activated MAPK pathway in myocardial ischemia/reperfusion injury. Life. Sci., 2015, 122: 15- 25. DOI: https://doi.org/10.1016/j.lfs.2014.11.014
- Shu Z. ; Yang Y. ; Yang L. ; et al . Cardioprotective effects of dihydroquercetin against ischemia reperfusion injury by inhibiting oxidative stress and endoplasmic reticulum stress-induced apoptosis via the PI3K/Akt pathway. Food. Funct., 2019, 10(1): 203-215. DOI: https://doi.org/10.1039/C8FO01256C
- Marks A . R. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J. Clin. Invest., 2013, 123( 1): 46- 52. DOI: https://doi.org/10.1172/JCI62834
- Balcazar D. ; Regge V. ; Santalla M. ; et al . SERCA is critical to control the Bowditch effect in the heart. Sci. Rep., 2018, 8(1): 12447. DOI: https://doi.org/10.1038/s41598-018-30638-9
- Zhu S. ; Xu T. ; Luo Y. ; et al . Luteolin enhances sarcoplasmic reticulum Ca 2+-ATPase activity through p38 MAPK signaling thus improving rat cardiac function after ischemia/reperfusion . Cell. Physiol. Biochem., 2017, 41(3): 999-1010. DOI: https://doi.org/10.1159/000460837
- Chen S. ; Li S . The Na +/Ca² + exchanger in cardiac ischemia/reperfusion injury . Med. Sci. Monit., 2012, 18(11): RA161- 165 DOI: https://doi.org/10.12659/MSM.883533
- Murphy E. ; Steenbergen C . Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev., 2008, 88(2): 581-609. DOI: https://doi.org/10.1152/physrev.00024.2007
- Aghaei M. ; Motallebnezhad M. ; Ghorghanlu S. ; et al . Targeting autophagy in cardiac ischemia/reperfusion injury: A novel therapeutic strategy. J. Cell. Physiol., 2019, 234(10): 16768-16778. DOI: https://doi.org/10.1002/jcp.28345
- Mozaffari M S. ; Liu J Y. ; Abebe W. ; et al . Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am. J. Cardiovasc. Dis., 2013, 3(4): 180-196.
- Haworth R. A. ; Hunter D . R. The Ca 2+-induced membrane transition in mitochondria . II. Nature of the Ca 2+ trigger site. Arch. Biochem. Biophys. , 1979, 195(2): 460-467. DOI: https://doi.org/10.1016/0003-9861(79)90372-2
- Hunter D. R. ; Haworth R . A. The Ca 2+-induced membrane transition in mitochondria . I. The protective mechanisms. Arch. Biochem. Biophys., 1979 195(2): 453-459. DOI: https://doi.org/10.1016/0003-9861(79)90371-0
- Santulli G. ; Xie W. ; Reiken S. R. ; et al . Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA., 2015, 112(36): 11389-11394. DOI: https://doi.org/10.1073/pnas.1513047112
- Fieni F. ; Johnson D E. ; Hudmon A. ; et al . Mitochondrial Ca 2+ uniporter and CaMKII in heart . Nature, 2014, 513(7519): E1-2. DOI: https://doi.org/10.1038/nature13626
- Verkhratsky A. ; Parpura V . Calcium signalling and calcium channels: evolution and general principles. Eur. J. Pharmacol., 2014, 739: 1-3. DOI: https://doi.org/10.1016/j.ejphar.2013.11.013
- Li S. ; Chen G. ; Li R . A. Calcium signalling of human pluripotent stem cell-derived cardiomyocytes. J. Physiol., 2013, 591(21): 5279-5290. DOI: https://doi.org/10.1113/jphysiol.2013.256495
- Yin B. ; Hou X. W. ; Lu M . L. Astragaloside IV attenuates myocardial ischemia/reperfusion injury in rats via inhibition of calcium-sensing receptor-mediated apoptotic signaling pathways. Acta. Pharmacol. Sin., 2019, 40(5): 599-607. DOI: https://doi.org/10.1038/s41401-018-0082-y
- Ye J. ; Wang R. ; Wang M. ; et al . Hydroxysafflor Yellow A ameliorates myocardial ischemia/reperfusion injury by suppressing calcium overload and apoptosis. Oxid. Med. Cell. Longev., 2021, 2021: 6643615. DOI: https://doi.org/10.1155/2021/6643615
- Wang R. ; Yang M. ; Wang M. ; et al . Total saponins of aralia elata (Miq) seem alleviate calcium homeostasis imbalance and endoplasmic reticulum stress-related apoptosis induced by myocardial ischemia/reperfusion injury. Cell. Physiol. Biochem., 2018, 50(1): 28-40. DOI: https://doi.org/10.1159/000493954
- Bou Teen. D. ; Kaludercic N. ; Weissman D. ; et al . Mitochondrial ROS and mitochondria-targeted antioxidants in the aged heart. Free. Radic. Biol. Med., 2021, 167: 109-124. DOI: https://doi.org/10.1016/j.freeradbiomed.2021.02.043
- Lesnefsky E. J. ; Chen Q. ; Tandler B. ; et al . Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu. Rev. Pharmacol. Toxicol., 2017, 57: 535-565. DOI: https://doi.org/10.1146/annurev-pharmtox-010715-103335
- Kwong J. Q. ; Molkentin J. D. ; Physiological and pathological roles of the mitochondrial permeability transition pore in the heart . Cell. Metab., 2015, 21(2): 206- 214. DOI: https://doi.org/10.1016/j.cmet.2014.12.001
- Lesnefsky E. J. ; Chen Q. ; Hoppel C . L. Mitochondrial metabolism in aging heart. Circ. Res., 2016, 118( 10): 1593- 1611. DOI: https://doi.org/10.1161/CIRCRESAHA.116.307505
- Ong S . B,; Hall A. R.; Hausenloy D. J.; Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox. Signal., 2013, 19( 4): 400- 414. DOI: https://doi.org/10.1089/ars.2012.4777
- Mui D.; Zhang Y.; Mitochondrial scenario: roles of mitochondrial dynamics in acute myocardial ischemia/reperfusion injury. J. Recept. Signal. Transduct. Res., 2021, 41(1): 1- 5. DOI: https://doi.org/10.1080/10799893.2020.1784938
- Chang J. C. ; Lien C. F. ; Lee W. S. ; et al . Intermittent hypoxia prevents myocardial mitochondrial Ca 2+ overload and cell death during ischemia/reperfusion: the role of reactive oxygen species . Cells, 2019, 8(6): 564 DOI: https://doi.org/10.3390/cells8060564
- Matsuda N. ; Sato S. ; Shiba K. ; et al . PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell. Biol., 2010, 189(2): 211-221. DOI: https://doi.org/10.1083/jcb.200910140
- Twig G. ; Elorza A. ; Molina A. J. ; et al . Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO. J., 2008, 27(2): 433-446. DOI: https://doi.org/10.1038/sj.emboj.7601963
- Twig G. ; Hyde B. ; Shirihai O . S. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta., 2008, 1777(9): 1092-1097. DOI: https://doi.org/10.1016/j.bbabio.2008.05.001
- Shi X. ; Li Y. ; Wang Y. ; et al . Pharmacological postconditioning with sappanone A ameliorates myocardial ischemia reperfusion injury and mitochondrial dysfunction via AMPK-mediated mitochondrial quality control. Toxicol. Appl. Pharmacol., 2021, 427: 115668. DOI: https://doi.org/10.1016/j.taap.2021.115668
- Wu B. ; Feng J. Y. ; Yu L. M. ; et al . Icariin protects cardiomyocytes against ischaemia/reperfusion injury by attenuating sirtuin 1-dependent mitochondrial oxidative damage. Br. J. Pharmacol., 2018, 175(21): 4137-4153. DOI: https://doi.org/10.1111/bph.14457
- Iurlaro R. ; Pinedo Munoz . C. Cell death induced by endoplasmic reticulum stress. FEBS. J., 2016, 283(14): 2640-2652. DOI: https://doi.org/10.1111/febs.13598
- Liang W. L. ; Cai M. R. ; Zhang M. Q. ; et al . Chinese herbal medicine alleviates myocardial ischemia/reperfusion injury by regulating endoplasmic reticulum stress. Evid. Based. Complement. Alternat. Med., 2021, 2021: 4963346. DOI: https://doi.org/10.1155/2021/4963346
- Liu M. Q.; Chen Z.; Chen L. X.; Endoplasmic reticulum stress: a novel mechanism and therapeutic target for cardiovascular diseases. Acta. Pharmacol. Sin., 2016, 37(4): 425-443. DOI: https://doi.org/10.1038/aps.2015.145
- Yang Y. ; Zhou Q. ; Gao A. ; et al . Endoplasmic reticulum stress and focused drug discovery in cardiovascular disease. Clin. Chim. Acta.; 2020, 504: 125-137. DOI: https://doi.org/10.1016/j.cca.2020.01.031
- Guan B. J. ; Krokowski D. ; Majumder M. ; et al . Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2alpha. J. Biol. Chem., 2014, 289(18): 12593-12611. DOI: https://doi.org/10.1074/jbc.M113.543215
- Lynch J. M. ; Maillet M. ; Vanhoutte D. ; et al . A thrombospondin-dependent pathway for a protective ER stress response. Cell, 2012, 149(6): 1257-1268. DOI: https://doi.org/10.1016/j.cell.2012.03.050
- Bertolotti A. ; Zhang Y. ; Hendershot L. M. ; et al . Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell. Biol., 2000, 2(6): 326-332. DOI: https://doi.org/10.1038/35014014
- Hetz C. ; Papa F . R. The unfolded protein response and cell fate control. Mol. Cell, 2018, 69( 2): 169- 181. DOI: https://doi.org/10.1016/j.molcel.2017.06.017
- Harding H. P. ; Calfon M. ; Urano F. ; et al . Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol., 2002, 18: 575-599. DOI: https://doi.org/10.1146/annurev.cellbio.18.011402.160624
- Ye J. ; Rawson R. B. ; Komuro R. ; et al . ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell, 2000, 6(6): 1355-1364. DOI: https://doi.org/10.1016/S1097-2765(00)00133-7
- Hu H. ; Tian M. ; Ding C. ; et al . The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol., 2018, 9: 3083. DOI: https://doi.org/10.3389/fimmu.2018.03083
- Walter P. ; Ron D . The unfolded protein response: from stress pathway to homeostatic regulation. Science, 2011, 334(6059): 1081-1086. DOI: https://doi.org/10.1126/science.1209038
- Malhotra J. D. ; Kaufman R . J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword?. Antioxid. Redox Signal., 2007, 9(12): 2277-2293. DOI: https://doi.org/10.1089/ars.2007.1782
- Tabas I. ; Ron D. ; Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress . Nat. Cell Biol., 2011, 13(3): 184-90. DOI: https://doi.org/10.1038/ncb0311-184
- Win S. ; Than T. A. ; Fernandez . Checa. J. C.; et al. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis., 2014, 5: e989. DOI: https://doi.org/10.1038/cddis.2013.522
- Chami M. ; Oules B. ; Szabadkai G. ; et al . Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell, 2008, 32( 5): 641-651. DOI: https://doi.org/10.1016/j.molcel.2008.11.014
- Moore C. E. ; Omikorede O. ; Gomez E. ; et al . PERK activation at low glucose concentration is mediated by SERCA pump inhibition and confers preemptive cytoprotection to pancreatic beta-cells. Mol. Endocrinol., 2011, 25(2): 315-326. DOI: https://doi.org/10.1210/me.2010-0309
- Rainbolt T. K. ; Saunders J. M. ; Wiseman R . L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab., 2014, 25(10): 528-537. DOI: https://doi.org/10.1016/j.tem.2014.06.007
- Verfaillie T. ; Rubio N. ; Garg A. D. ; et al . PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ., 2012, 19(11): 1880-1891. DOI: https://doi.org/10.1038/cdd.2012.74
- Kolattukudy P. E. ; Niu J . Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ. Res., 2012, 110(1): 174-189. DOI: https://doi.org/10.1161/CIRCRESAHA.111.243212
- Deng J. ; Lu P. D. ; Zhang Y. ; et al . Translational repression mediates activation of nuclear factor kappa B by phosphorylation of translation initiation factor 2. Mol. Cell. Biol., 2004, 24(23): 10161-10168. DOI: https://doi.org/10.1128/MCB.24.23.10161-10168.2004
- Cui Y. ; Wang Y. ; Liu G . Protective effect of Barbaloin in a rat model of myocardial ischemia reperfusion injury through the regulation of the CNPY2‑PERK pathway. Int. J. Mol. Med., 2019, 43(5): 2015-2023. DOI: https://doi.org/10.3892/ijmm.2019.4123
- Yu Y. ; Xing N. ; Xu X. ; et al . Tournefolic acid B, derived from Clinopodium chinense (Benth.) Kuntze, protects against myocardial ischemia/reperfusion injury by inhibiting endoplasmic reticulum stress-regulated apoptosis via PI3K/AKT pathways. Phytomedicine, 2019, 52: 178-186. DOI: https://doi.org/10.1016/j.phymed.2018.09.168
- Algoet M. ; Janssens S. ; Himmelreich U. ; et al . Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc. Med., 2022. DOI: https://doi.org/10.1016/j.tcm.2022.02.005
- Zhang D. ; Wu H. ; Liu D. ; et al . Research progress on the mechanism and treatment of inflammatory response in myocardial ischemia-reperfusion injury. Heart Surg. Forum., 2022, 25(3): E462-E468. DOI: https://doi.org/10.1532/hsf.4725
- Kelley N. ; Jeltema D. ; Duan Y. ; et al . The NLRP3 Inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci., 2019, 20( 13). 3328 DOI: https://doi.org/10.3390/ijms20133328
- Wang D. ; Lv L. ; Xu Y. ; et al . Cardioprotection of Panax Notoginseng saponins against acute myocardial infarction and heart failure through inducing autophagy. Biomed. Pharmacother., 2021, 136: 111287. DOI: https://doi.org/10.1016/j.biopha.2021.111287
- Takeuchi O. ; Akira S . Pattern recognition receptors and inflammation. Cell, 2010, 140(6): 805-820. DOI: https://doi.org/10.1016/j.cell.2010.01.022
- Zheng D. ; Liwinski T. ; Elinav E . Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov., 2020, 6: 36. DOI: https://doi.org/10.1038/s41421-020-0167-x
- Shen S. ; Wang Z. ; Sun H. ; et al . Role of NLRP3 inflammasome in myocardial ischemia-reperfusion injury and ventricular remodeling. Med. Sci. Monit., 2022, 28: e934255. DOI: https://doi.org/10.12659/MSM.934255
- Wang P. F. ; Xiong X. Y. ; Chen J. ; et al . Function and mechanism of toll-like receptors in cerebral ischemic tolerance: from preconditioning to treatment. J. Neuroinflammation, 2015, 12: 80. DOI: https://doi.org/10.1186/s12974-015-0301-0
- Gurung P. ; Anand P. K. ; Malireddi R. K. ; et al . FADD and caspase-8 mediate priming and activation of the canonical and noncanonical NLRP3 inflammasomes. J. Immunol., 2014, 192(4): 1835-1846. DOI: https://doi.org/10.4049/jimmunol.1302839
- Wang Z. ; Zhang S. ; Xiao Y. ; et al . NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev., 2020, 2020: 4063562. DOI: https://doi.org/10.1155/2020/4063562
- Chen L. ; Liu P. ; Feng X. ; et al . Salidroside suppressing LPS-induced myocardial injury by inhibiting ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J. Cell. Mol. Med., 2017, 21(12): 3178-3189. DOI: https://doi.org/10.1111/jcmm.12871
- Fan Q. ; Tao R. ; Zhang H. ; et al . Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation, 2019, 139(5): 663-678. DOI: https://doi.org/10.1161/CIRCULATIONAHA.118.036044
- Huang X. W. ; Pan M. D. ; Du P. H. ; et al . Arginase-2 protects myocardial ischemia-reperfusion injury via NF-κB/TNF-α pathway. Eur. Rev. Med. Pharmacol. Sci., 2018, 22(19): 6529-6537.
- Zhou R. ; Yazdi A. S. ; Menu P. ; et al . A role for mitochondria in NLRP3 inflammasome activation. Nature, 2011, 469(7329): 221-225. DOI: https://doi.org/10.1038/nature09663
- Li D. ; Yang S. ; Xing Y. ; et al . Novel insights and current evidence for mechanisms of atherosclerosis: Mitochondrial dynamics as a potential therapeutic target. Front. Cell Dev. Biol., 2021, 9: 673839. DOI: https://doi.org/10.3389/fcell.2021.673839
- Lee G. S. ; Subramanian N. ; Kim A. I. ; et al . The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca 2+ and cAMP . Nature, 2012, 492(7427): 123-127. DOI: https://doi.org/10.1038/nature11588
- Peng L. ; Lei Z. ; Rao Z. ; et al . Cardioprotective activity of ethyl acetate extract of Cinnamomi Ramulus against myocardial ischemia/reperfusion injury in rats via inhibiting NLRP3 inflammasome activation and pyroptosis. Phytomedicine, 2021, 93: 153798. DOI: https://doi.org/10.1016/j.phymed.2021.153798
- Wang F. ; Gao Q. ; Yang J. ; et al . Artemisinin suppresses myocardial ischemia-reperfusion injury via NLRP3 inflammasome mechanism. Mol. Cell. Biochem., 2020, 474(1-2): 171-180. DOI: https://doi.org/10.1007/s11010-020-03842-3
- Chen Z. ; Wu J. ; Li S. ; et al . Inhibition of myocardial cell apoptosis is important mechanism for Ginsenoside in the limitation of myocardial ischemia/reperfusion injury. Front. Pharmacol., 2022, 13: 806216. DOI: https://doi.org/10.3389/fphar.2022.806216
- Gottlieb R. A. ; Engler R. L. ; Apoptosis in myocardial ischemia-reperfusion . Ann. N. Y. Acad. Sci, 1999, 874: 412-426. DOI: https://doi.org/10.1111/j.1749-6632.1999.tb09255.x
- Dong Y. ; Chen H. ; Gao J. ; et al . Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol., 2019, 136: 27-41. DOI: https://doi.org/10.1016/j.yjmcc.2019.09.001
- Green D R . The Death Receptor Pathway of Apoptosis. Cold Spring Harb. Perspect. Biol., 2022, 14( 2): a041053. DOI: https://doi.org/10.1101/cshperspect.a041053
- He J. ; Liu D. ; Zhao L. ; et al . Myocardial ischemia/reperfusion injury: Mechanisms of injury and implications for management (Review). Exp. Ther. Med., 2022, 23(6): 430. DOI: https://doi.org/10.3892/etm.2022.11357
- Gupta S. ; Kass G. E. ; Szegezdi E. ; et al . The mitochondrial death pathway: a promising therapeutic target in diseases. J. Cell. Mol. Med., 2009, 13(6): 1004-1033. DOI: https://doi.org/10.1111/j.1582-4934.2009.00697.x
- Korshunova . A. Y.; Blagonravov. M. L.; Neborak. E. V.; et al. BCL2‑regulated apoptotic process in myocardial ischemia‑reperfusion injury (Review). Int. J. Mol. Med., 2021, 47(1): 23-36. DOI: https://doi.org/10.3892/ijmm.2020.4781
- Su X. ; Zhou M. ; Li Y. ; et al . Mitochondrial damage in myocardial ischemia/reperfusion injury and application of natural plant products. Oxid. Med. Cell. Longev., 2022, 2022: 8726564. DOI: https://doi.org/10.1155/2022/8726564
- Miyazaki Y. ; Kaikita K. ; Endo M. ; et al . C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler. Thromb. Vasc. Biol., 2011, 31(5): 1124-1132. DOI: https://doi.org/10.1161/ATVBAHA.111.224519
- Morishima N. ; Nakanishi K. ; Tsuchiya K. ; et al . Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem., 2004, 279(48): 50375-50381. DOI: https://doi.org/10.1074/jbc.M408493200
- Yang T. R. ; Zhang T. ; Mu N. H. ; et al . Resina draconis inhibits the endoplasmic-reticulum-induced apoptosis of myocardial cells via regulating miR-423-3p/ERK signaling pathway in a tree shrew myocardial ischemia–reperfusion model. J. Biosci., 2019, 44(2). DOI: https://doi.org/10.1007/s12038-019-9872-8
- Saha S. ; Panigrahi D. P. ; Patil S . et al . Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother, 2018, 104: 485-495. DOI: https://doi.org/10.1016/j.biopha.2018.05.007
- Shi B. ; Ma M. ; Zheng Y. ; et al . mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J. Cell. Physiol., 2019, 234( 8): 12562-12568. DOI: https://doi.org/10.1002/jcp.28125
- Daniels L. J. ; Varma U. ; Annandale M. ; et al . Myocardial energy stress, autophagy induction, and cardiomyocyte functional responses. Antioxid. Redox Signal., 2019, 31( 6): 472-486. DOI: https://doi.org/10.1089/ars.2018.7650
- Petrovski G. ; Das S. ; Juhasz B. ; et al . Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxid. Redox Signal., 2011, 14( 11): 2191-2200. DOI: https://doi.org/10.1089/ars.2010.3486
- Ma S. ; Wang Y. ; Chen Y. ; et al . The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta, 2015, 1852( 2): 271-276. DOI: https://doi.org/10.1016/j.bbadis.2014.05.010
- Matsui Y. ; Takagi H. ; Qu X. ; et al . Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res., 2007, 100( 6): 914-922. DOI: https://doi.org/10.1161/01.RES.0000261924.76669.36
- Zhang D. ; He Y. ; Ye X. ; et al . Activation of autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome activation and attenuates myocardial ischemia-reperfusion injury in diabetic rats. J. Diabetes Investig., 2020, 11( 5): 1126-1136. DOI: https://doi.org/10.1111/jdi.13235
- Wang Q. ; Zhang H. ; Wang B. ; et al . beta adrenergic receptor/cAMP/PKA signaling contributes to the intracellular Ca(2+) release by tentacle extract from the jellyfish Cyanea capillata. BMC Pharmacol. Toxicol., 2017, 18( 1): 60. DOI: https://doi.org/10.1186/s40360-017-0167-0
- Brady N. R. ; Hamacher Brady. A. ; Yuan H. ; et al . The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J., 2007, 274( 12): 3184-3197. DOI: https://doi.org/10.1111/j.1742-4658.2007.05849.x
- Hariharan N. ; Zhai P. ; Sadoshima J . Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal., 2011, 14( 11): 2179-2190. DOI: https://doi.org/10.1089/ars.2010.3488
- Ma X. ; Liu H. ; Foyil S. R. ; et al . Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy, 2012, 8( 9): 1394-1396. DOI: https://doi.org/10.4161/auto.21036
- Wang D. ; Yu W. ; Liu Y. ; et al . Roles of autophagy in ischemic heart diseases and the modulatory effects of chinese herbal medicine. Am. J. Chin. Med., 2017, 45( 7): 1401-1419. DOI: https://doi.org/10.1142/S0192415X17500768
- Fu S. ; Chen L. ; Wu Y. ; et al . Gastrodin pretreatment alleviates myocardial ischemia/reperfusion injury through promoting autophagic flux. Biochem. Biophys. Res. Commun., 2018, 503( 4): 2421-2428. DOI: https://doi.org/10.1016/j.bbrc.2018.06.171
- Qin G.W. ; Lu P. ; Peng L. ; et al . Ginsenoside Rb1 Inhibits Cardiomyocyte Autophagy via PI3K/Akt/mTOR Signaling Pathway and Reduces Myocardial Ischemia/Reperfusion Injury. Am. J. Chin. Med., 2021, 49( 8): 1913-1927. DOI: https://doi.org/10.1142/S0192415X21500907
- Gonzalez Vallinas. M. ; Gonzalez Castejon. M. ; Rodriguez Casado. A. ; et al . Dietary phytochemicals in cancer prevention and therapy: a complementary approach with promising perspectives. Nutr. Rev., 2013, 71( 9): 585-599. DOI: https://doi.org/10.1111/nure.12051
- Araujo J. R. ; Goncalves P. ; Martel F . Chemopreventive effect of dietary polyphenols in colorectal cancer cell lines. Nutr. Res., 2011, 31( 2): 77-87. DOI: https://doi.org/10.1016/j.nutres.2011.01.006
- Li A. N. ; Li S. ; Zhang Y. J. ; et al . Resources and biological activities of natural polyphenols. Nutrients, 2014, 6( 12): 6020-6047. DOI: https://doi.org/10.3390/nu6126020
- Brglez Mojzer. E. ; Knez Hrncic. M. ; Skerget M. ; et al . Polyphenols: Extraction methods, antioxidative action, bioavailability and anticarcinogenic effects. Molecules, 2016, 21(7): 901. DOI: https://doi.org/10.3390/molecules21070901
- Rajendran P. ; Abdelsalam S. A. ; Renu K. ; et al . Polyphenols as potent epigenetics Agents for Cancer. Int. J. Mol. Sci., 2022, 23( 19): 11712. DOI: https://doi.org/10.3390/ijms231911712
- Chen Y. ; Ba L. ; Huang W. ; et al . Role of carvacrol in cardioprotection against myocardial ischemia/reperfusion injury in rats through activation of MAPK/ERK and Akt/eNOS signaling pathways. Eur. J. Pharmacol., 2017, 796: 90-100. DOI: https://doi.org/10.1016/j.ejphar.2016.11.053
- Tian Y. ; Du Y. Y. ; Shang H. ; et al . Calenduloside E analogues protecting H9c2 cardiomyocytes against H 2O 2-induced apoptosis: design, synthesis and biological evaluation . Front. Pharmacol., 2017, 8: 862. DOI: https://doi.org/10.3389/fphar.2017.00862
- Yang H. ; Xue W. ; Ding C. ; et al . Vitexin mitigates myocardial ischemia/reperfusion injury in rats by regulating mitochondrial dysfunction via Epac1-Rap1 signaling. Oxid. Med. Cell. Longev., 2021, 2021: 9921982. DOI: https://doi.org/10.1155/2021/9921982
- Wang Y. ; Che J. ; Zhao H. ; et al . Platycodin D inhibits oxidative stress and apoptosis in H9c2 cardiomyocytes following hypoxia/reoxygenation injury. Biochem. Biophys. Res. Commun., 2018, 503( 4): 3219-3224. DOI: https://doi.org/10.1016/j.bbrc.2018.08.129
- Chen J. ; Ko K . M. Ursolic-Acid-Enriched Herba Cynomorii extract protects against oxidant injury in H9c2 cells and rat myocardium by increasing mitochondrial ATP generation capacity and enhancing cellular glutathione redox cycling, possibly through mitochondrial uncoupling. Evid. Based Complement. Alternat. Med., 2013, 2013: 924128. DOI: https://doi.org/10.1155/2013/924128
- Sun W. ; Lu H. ; Lyu L. ; et al . Gastrodin ameliorates microvascular reperfusion injury-induced pyroptosis by regulating the NLRP3/caspase-1 pathway. J. Physiol. Biochem., 2019, 75( 4): 531-547. DOI: https://doi.org/10.1007/s13105-019-00702-7
- Xu T. ; Qin G. ; Jiang W. ; et al . 6-Gingerol protects heart by suppressing myocardial ischemia/reperfusion induced inflammation via the PI3K/Akt-dependent mechanism in rats. Evid. Based Complement. Alternat. Med., 2018, 2018: 6209679. DOI: https://doi.org/10.1155/2018/6209679
- Tong S. ; Zhang L. ; Joseph J. ; et al . Celastrol pretreatment attenuates rat myocardial ischemia/ reperfusion injury by inhibiting high mobility group box 1 protein expression via the PI3K/Akt pathway. Biochem. Biophys. Res. Commun., 2018, 497( 3): 843-849. DOI: https://doi.org/10.1016/j.bbrc.2018.02.121
- Ye B. ; Chen X. ; Dai S. ; et al . Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug. Des. Devel. Ther., 2019, 13: 975-990. DOI: https://doi.org/10.2147/DDDT.S195412
- Wei W. ; Peng J. ; Li J . Curcumin attenuates hypoxia/reoxygenation‑induced myocardial injury. Mol. Med. Rep., 2019, 20( 6): 4821-4830. DOI: https://doi.org/10.3892/mmr.2019.10742
- Jo W. ; Min B. S. ; Yang H. Y. ; et al . Sappanone A prevents left ventricular dysfunction in a rat myocardial ischemia reperfusion injury model. Int. J. Mol. Sci., 2020, 21(18): 6935. DOI: https://doi.org/10.3390/ijms21186935
- Deng Y. ; Chen G. ; Ye M. ; et al . Bifunctional supramolecular hydrogel alleviates myocardial ischemia/reperfusion injury by inhibiting autophagy and apoptosis. J. Biomed. Nanotechnol, 2018, 14( 8): 1458-1470. DOI: https://doi.org/10.1166/jbn.2018.2582
- Osbourn A. ; Goss R. J. ; Field R . A. The saponins: polar isoprenoids with important and diverse biological activities. Nat. Prod. Rep., 2011, 28( 7): 1261-1268. DOI: https://doi.org/10.1039/c1np00015b
- Güçlü Üstündağ. Ö. ; Mazza G . Saponins: properties, applications and processing. Crit. Rev. Food. Sci. Nutr., 2007, 47( 3): 231-258. DOI: https://doi.org/10.1080/10408390600698197
- Su H. F. ; Shaker S. ; Kuang Y. ; et al . Phytochemistry and cardiovascular protective effects of Huang-Qi (Astragali Radix). Med. Res. Rev., 2021, 41( 4): 1999-2038. DOI: https://doi.org/10.1002/med.21785
- Al-Kuraishy H. M. ; Hussaniy Al . H. A.; Al Gareeb. A. I.; et al. Combination of Panax ginseng C. A. Mey and Febuxostat boasted cardioprotective effects against doxorubicin-induced acute cardiotoxicity in rats. Front. Pharmacol., 2022, 13: 905828. DOI: https://doi.org/10.3389/fphar.2022.905828
- Wang Q. ; Mu R. F. ; Liu X. ; et al . Steaming changes the composition of saponins of panax notoginseng (Burk.) F. H. Chen that function in treatment of hyperlipidemia and obesity. J. Agric. Food. Chem., 2020, 68( 17): 4865-4875. DOI: https://doi.org/10.1021/acs.jafc.0c00746
- Szczuka D. ; Nowak A. ; Zaklos Szyda. M. ; et al . American Ginseng (Panax quinquefolium L.) as a source of bioactive phytochemicals with pro-health properties. Nutrients, 2019, 11( 5): 1041. DOI: https://doi.org/10.3390/nu11051041
- Zhou P. ; Xie W. ; Luo Y. ; et al . Protective effects of total Saponins of Aralia elata (Miq.) on endothelial cell injury induced by TNF-alpha via modulation of the PI3K/Akt and NF-kappaB signalling pathways. Int. J. Mol. Sci., 2018, 20( 1): 36. DOI: https://doi.org/10.3390/ijms20010036
- Zhang L. ; Yong Y. Y. ; Deng L. ; et al . Therapeutic potential of polygala saponins in neurological diseases. Phytomedicine, 2023, 108: 154483. DOI: https://doi.org/10.1016/j.phymed.2022.154483
- Zhong J. ; Tan L. ; Chen M. ; et al . Pharmacological activities and molecular mechanisms of Pulsatilla saponins. Chin. Med., 2022, 17( 1): 59. DOI: https://doi.org/10.1186/s13020-022-00613-8
- Yi C. ; Song M. ; Sun L. ; et al . Asiatic Acid alleviates myocardial ischemia-Reperfusion Injury by Inhibiting the ROS-Mediated Mitochondria-Dependent Apoptosis Pathway. Oxid. Med. Cell. Longev., 2022, 2022: 3267450. DOI: https://doi.org/10.1155/2022/3267450
- Huang X. ; Wang Y. ; Wang Y. ; et al . Ophiopogonin D reduces myocardial ischemia-reperfusion injury via upregulating CYP2J3/EETs in Rats. Cell. Physiol. Biochem., 2018, 49( 4): 1646-1658. DOI: https://doi.org/10.1159/000493500
- Cui Y. C. ; Pan C. S. ; Yan L. ; et al . Ginsenoside Rb1 protects against ischemia/reperfusion-induced myocardial injury via energy metabolism regulation mediated by RhoA signaling pathway. Sci. Rep., 2017, 7: 44579. DOI: https://doi.org/10.1038/srep44579
- Liu X. W. ; Lu M. K. ; Zhong H. T. ; et al . Panax Notoginseng Saponins Attenuate Myocardial Ischemia-Reperfusion Injury Through the HIF-1α/BNIP3 Pathway of Autophagy. J. Cardiovasc. Pharmacol., 2019, 73( 2): 92-99. DOI: https://doi.org/10.1097/FJC.0000000000000640
- Tian L. ; Cao W. ; Yue R. ; et al . Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. J. Pharmacol. Sci., 2019, 139( 4): 352-360. DOI: https://doi.org/10.1016/j.jphs.2019.02.008
- Zhao Y. ; Guo Y. ; Chen Y. ; et al . Curculigoside attenuates myocardial ischemia‑reperfusion injury by inhibiting the opening of the mitochondrial permeability transition pore. Int. J. Mol. Med., 2020, 45( 5): 1514-1524. DOI: https://doi.org/10.3892/ijmm.2020.4513
- Yu Y. ; Sun G. ; Luo Y. ; et al . Cardioprotective effects of Notoginsenoside R1 against ischemia/reperfusion injuries by regulating oxidative stress- and endoplasmic reticulum stress- related signaling pathways. Sci. Rep., 2016, 6: 21730. DOI: https://doi.org/10.1038/srep21730
- Du Y. ; Wang M. ; Liu X. ; et al . Araloside C prevents hypoxia/reoxygenation-induced endoplasmic reticulum stress via increasing heat shock protein 90 in H9c2 cardiomyocytes. Front. Pharmacol., 2018, 9: 180. DOI: https://doi.org/10.3389/fphar.2018.00180
- Christianson D . W. Structural and chemical biology of terpenoid cyclases. Chem. Rev., 2017, 117( 17): 11570- 11648. DOI: https://doi.org/10.1021/acs.chemrev.7b00287
- Liao P. ; Hemmerlin A. ; Bach T J. ; et al . The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol. Adv., 2016, 34( 5): 697-713. DOI: https://doi.org/10.1016/j.biotechadv.2016.03.005
- Hemmerlin A. ; Harwood J. L. ; Bach T . J. A raison d'etre for two distinct pathways in the early steps of plant isoprenoid biosynthesis?. Prog. Lipid. Res., 2012, 51( 2): 95-148. DOI: https://doi.org/10.1016/j.plipres.2011.12.001
- Bicchi C. ; Appendino G. ; Cordero C. ; et al . HPLC-UV and HPLC-positive-ESI-MS analysis of the diterpenoid fraction from caper spurge (Euphorbia lathyris) seed oil. Phytochem. Anal., 2001, 12( 4): 255-262. DOI: https://doi.org/10.1002/pca.592
- Zhang L. L. ; Xu W. ; Xu Y. L. ; et al . Therapeutic potential of Rhizoma Alismatis: a review on ethnomedicinal application, phytochemistry, pharmacology, and toxicology. Ann. N. Y. Acad. Sci., 2017, 1401( 1): 90-101. DOI: https://doi.org/10.1111/nyas.13381
- Orgah J. O. ; He S. ; Wang Y. ; et al . Pharmacological potential of the combination of Salvia miltiorrhiza (Danshen) and Carthamus tinctorius (Honghua) for diabetes mellitus and its cardiovascular complications. Pharmacol. Res., 2020, 153: 104654. DOI: https://doi.org/10.1016/j.phrs.2020.104654
- Suh K. S. ; Chon S. ; Jung W. W. ; et al . Crocin attenuates methylglyoxal-induced osteoclast dysfunction by regulating glyoxalase, oxidative stress, and mitochondrial function. Food. Chem. Toxicol., 2019, 124: 367-373. DOI: https://doi.org/10.1016/j.fct.2018.12.031
- Thummuri D. ; Guntuku L. ; Challa V. S. ; et al . Abietic acid attenuates RANKL induced osteoclastogenesis and inflammation associated osteolysis by inhibiting the NF-KB and MAPK signaling. J. Cell. Physiol., 2018, 234( 1): 443-453. DOI: https://doi.org/10.1002/jcp.26575
- Chen L. ; Wei L. ; Yu Q. ; et al . Tanshinone IIA alleviates hypoxia/reoxygenation induced cardiomyocyte injury via lncRNA AK003290/miR-124-5p signaling. BMC Mol. Cell Biol., 2020, 21( 1): 20. DOI: https://doi.org/10.1186/s12860-020-00264-3
- Zhuo Y. ; Yuan R. ; Chen X. ; et al . Tanshinone I exerts cardiovascular protective effects in vivo and in vitro through inhibiting necroptosis via Akt/Nrf2 signaling pathway. Chin. Med., 2021, 16( 1): 48. DOI: https://doi.org/10.1186/s13020-021-00458-7
- Schlager S. ; Drager B . Exploiting plant alkaloids. Curr. Opin. Biotechnol., 2016, 37: 155-164. DOI: https://doi.org/10.1016/j.copbio.2015.12.003
- Zhang Y. ; Li M. ; Li X. ; et al . Isoquinoline Alkaloids and Indole Alkaloids attenuate aortic atherosclerosis in apolipoprotein E deficient mice: a systematic review and meta-analysis. Front. Pharmacol., 2018, 9: 602. DOI: https://doi.org/10.3389/fphar.2018.00602
- Liu W. ; Zhang Y. ; Zhu W. ; et al . Sinomenine inhibits the progression of rheumatoid arthritis by regulating the secretion of inflammatory cytokines and monocyte/macrophage subsets. Front. Immunol., 2018, 9: 2228. DOI: https://doi.org/10.3389/fimmu.2018.02228
- Ma Y. ; Yu W. ; Shrivastava A. ; et al . Sanguinarine inhibits pancreatic cancer stem cell characteristics by inducing oxidative stress and suppressing sonic hedgehog-Gli-Nanog pathway. Carcinogenesis, 2017, 38( 10): 1047-1056. DOI: https://doi.org/10.1093/carcin/bgx070
- Michael J . P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep., 2008, 25( 1): 139-65. DOI: https://doi.org/10.1039/B612166G
- Zhu N. ; Li J. ; Li Y. ; et al . Berberine protects against simulated ischemia/reperfusion injury-induced H9C2 cardiomyocytes apoptosis in vitro and myocardial ischemia/reperfusion-induced apoptosis in vivo by regulating the mitophagy-mediated HIF-1alpha/BNIP3 pathway. Front. Pharmacol., 2020, 11: 367. DOI: https://doi.org/10.3389/fphar.2020.00367
- Zhao G. L. ; Yu L. M. ; Gao W. L. ; et al . Berberine protects rat heart from ischemia/reperfusion injury via activating JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress. Acta. Pharmacol. Sin., 2016, 37( 3): 354- 67. DOI: https://doi.org/10.1038/aps.2015.136
- Wongcharoen W. ; Jai Aue S. ; Phrommintikul A. ; Effects of curcuminoids on frequency of acute myocardial infarction after coronary artery bypass grafting . Am. J. Cardiol., 2012, 110( 1): 40- 44. DOI: https://doi.org/10.1016/j.amjcard.2012.02.043
- Aslanabadi N. ; Entezari-Maleki T. ; Rezaee H. ; et al . Curcumin for the prevention of myocardial injury following elective percutaneous coronary intervention; a pilot randomized clinical trial. Eur. J. Pharmacol., 2019, 858: 172471. DOI: https://doi.org/10.1016/j.ejphar.2019.172471
- Asgary S. ; Soltani R. ; Daraei F. ; et al . The effect of lycopene on serum level of cardiac biomarkers in patients undergoing elective percutaneous coronary intervention: A randomized controlled clinical trial. ARYA Atheroscler., 2021, 17( 1): 1- 7.
- Aslanabadi N. ; Jafaripor I. ; Sadeghi S. ; et al . Effect of Vitamin D in the prevention of myocardial injury following elective percutaneous coronary intervention: A pilot randomized clinical trial. J. Clin. Pharmacol., 2018, 58( 2): 144- 151. DOI: https://doi.org/10.1002/jcph.989
- Zhu P C. ; Tong Q. ; Zhuang Z. ; et al . Ginkgolide B for myocardial ischemia/reperfusion injury: A preclinical systematic review and meta-analysis. Front. Physiol., 2019, 10: 1292. DOI: https://doi.org/10.3389/fphys.2019.01292
- Liu M. ; Ramponi C. ; Fan X. ; et al . Nanoparticle-based Drug Delivery System for Post Myocardial Infarction Management. Inter. J. Drug Discov. Pharmacol., 2022, 1( 1): 11. DOI: https://doi.org/10.53941/ijddp.v1i1.171


