Subcellular organellar contact sites, particularly those between mitochondria and the endoplasmic reticulum (MERCSs), play crucial roles in maintaining health. These specialized partitions facilitate vital communication between the organelles, regulating processes essential for cell function, including calcium balance, lipid biogenesis and transport, mitochondrial dynamics, and programmed cell death. Growing evidence shows that perturbation of MERCSs contributes significantly to various diseases, including neurodegenerative disorders like Alzheimer’s and Parkinson’s, metabolic issues, such as type 2 diabetes, heart conditions, and cancer. This review dives into this expanding field, exploring MERCSs as potential therapeutic targets. It provides a detailed overview of the proteins and processes that form and maintain MERCSs, highlighting how their disruption can lead to cellular dysfunction and disease. Additionally, it examines recent exciting breakthroughs in developing drugs and strategies that can manipulate MERCSs for clinical benefits. While challenges remain, this review emphasises the potential of MERCS-based therapies and outlines the critical research needed to move these treatments from the lab to the clinic.
- Open Access
- Review
Emerging and Novel Therapeutic Treatments Targeting Mitochondrial-Endoplasmic Reticulum Contact Sites in Metabolic and Vascular Disorders
- Richard M. Monaghan
Author Information
Received: 10 Apr 2024 | Revised: 05 May 2024 | Accepted: 07 May 2024 | Published: 06 Jun 2024
Abstract
Graphical Abstract
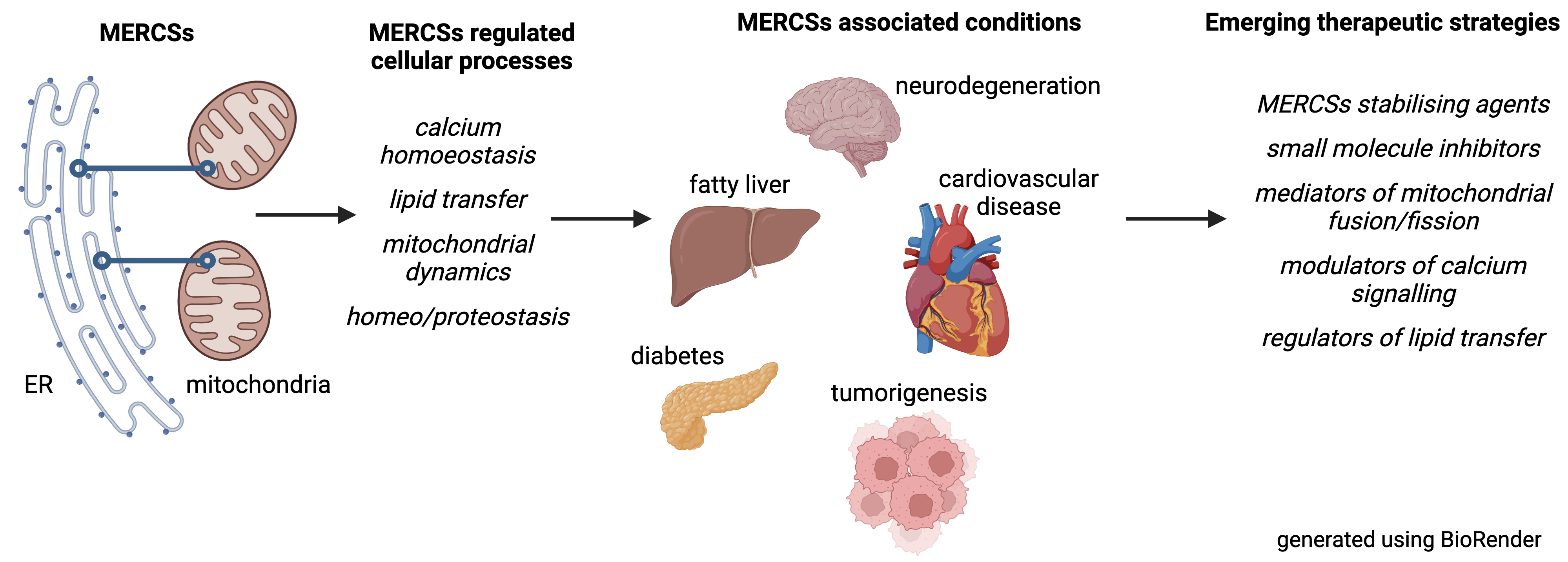
Keywords
MERCSs | neurodegeneration | calcium signaling | lipid metabolism | mitochondrial dynamics
References
- 1.Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; et al. Mitochondria-hubs for regulating cellular biochemistry: emerging concepts and networks. Open Biol. 2019, 9, 190126, https://doi.org/10.1098/rsob.190126.
- 2.Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94, https://doi.org/10.1007/s00018-015-2052-6.
- 3.Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201, https://doi.org/10.1101/cshperspect.a013201.
- 4.Sun, S.; Zhao, G.; Jia, M.; et al. Stay in touch with the endoplasmic reticulum. Sci. China Life Sci. 2024, 67, 230–257, https://doi.org/10.1007/s11427-023-2443-9.
- 5.Yuan, M.; Gong, M.; He, J.; et al. IP3R1/GRP75/VDAC1 complex mediates endoplasmic reticulum stress-mitochondrial oxidative stress in diabetic atrial remodeling. Redox Biol. 2022, 52, 102289, https://doi.org/10.1016/j.redox.2022.102289.
- 6.Filadi, R.; Leal, N.S.; Schreiner, B.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca(2+) Transfer. Curr Biol, 2018. 28, 369–382. https://doi.org/10.1016/j.cub.2017.12.047
- 7.D’Eletto, M.; Rossin, F.; Occhigrossi, L.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573‒3581, https://doi.org/10.1016/j.celrep.2018.11.094.
- 8.Matsuzaki, H.; Fujimoto, T.; Tanaka, M.; et al. Tespa1 is a novel component of mitochondria-associated endoplasmic reticulum membranes and affects mitochondrial calcium flux. Biochem. Biophys. Res. Commun. 2013, 433, 322–326, https://doi.org/10.1016/j.bbrc.2013.02.099.
- 9.Thoudam, T.; Ha, C.-M.; Leem, J.; et al. PDK4 Augments ER–Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 2018, 68, 571–586, https://doi.org/10.2337/db18-0363.
- 10.Ilacqua, N.; Sánchez-Álvarez, M.; Bachmann, M.; et al. Protein Localization at Mitochondria-ER Contact Sites in Basal and Stress Conditions. Front. Cell Dev. Biol. 2017, 5, 107, https://doi.org/10.3389/fcell.2017.00107.
- 11.Han, S.; Zhao, F.; Hsia, J.; et al. The role of Mfn2 in the structure and function of endoplasmic reticulum-mitochondrial tethering in vivo. J. Cell Sci. 2021, 134, jcs253443, https://doi.org/10.1242/jcs.253443.
- 12.Naón, D.; Hernández-Alvarez, M.I.; Shinjo, S.; et al. Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science 2023, 380, eadh9351, https://doi.org/10.1126/science.adh9351.
- 13.Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; et al. Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria. Am. J. Pathol. 2009, 175, 1810–1816, doi:10.2353/ajpath.2009.090219.
- 14.Contino, S.; Porporato, P.E.; Bird, M.; et al. Presenilin 2-Dependent Maintenance of Mitochondrial Oxidative Capacity and Morphology. Front. Physiol. 2017, 8, 796, https://doi.org/10.3389/fphys.2017.00796.
- 15.Filadi, R.; Greotti, E.; Turacchio, G.; et al. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238, https://doi.org/10.1016/j.celrep.2016.05.013.
- 16.Sugiura, A.; Nagashima, S.; Tokuyama, T.; et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34, https://doi.org/10.1016/j.molcel.2013.04.023.
- 17.Yamano, K.; Kinefuchi, H.; Kojima, W. Mitochondrial lipid dynamics regulated by MITOL-mediated ubiquitination. J. Biochem. 2023, 175, 217–219, https://doi.org/10.1093/jb/mvad117.
- 18.Wang, H.; Ju, D.; Kho, D.-H.; et al. The ubiquitin specific protease USP34 protects the ubiquitin ligase gp78 from proteasomal degradation. Biochem. Biophys. Res. Commun. 2019, 509, 348–353, https://doi.org/10.1016/j.bbrc.2018.12.141.
- 19.De Vos, K.J.; Morotz, G.M.; Stoica, R.; et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet, 2012, 21, 1299–1311.
- 20.Mórotz, G.M.; Martín-Guerrero, S.M.; Markovinovic, A.; et al. The PTPIP51 coiled-coil domain is important in VAPB binding, formation of ER-mitochondria contacts and IP3 receptor delivery of Ca(2+) to mitochondria. Front Cell Dev Biol. 2022. 10, 920947.
- 21.Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; et al. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2010, 30, 556–568, https://doi.org/10.1038/emboj.2010.346.
- 22.Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, eaaw1386, https://doi.org/10.1126/sciadv.aaw1386.
- 23.Guillén-Samander, A.; Leonzino, M.; Hanna, IVM.G.; et al. VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004, https://doi.org/10.1083/jcb.202010004
- 24.Hung, V.; Lam, S.S.; Udeshi, N.D., et al. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 2017, 6, e24463, https://doi.org/10.7554/eLife.24463
- 25.Doghman‐Bouguerra, M.; Granatiero, V.; Sbiera, S.; et al. FATE 1 antagonizes calcium‐ and drug‐induced apoptosis by uncoupling ER and mitochondria. Embo Rep. 2016, 17, 1264–1280, https://doi.org/10.15252/embr.201541504.
- 26.Liao, H.-Y.; Liao, B.; Zhang, H.-H. CISD2 plays a role in age-related diseases and cancer. Biomed. Pharmacother. 2021, 138, 111472, https://doi.org/10.1016/j.biopha.2021.111472.
- 27.Ilamathi, H.S.; Benhammouda, S.; Lounas, A.; et al. Contact sites between endoplasmic reticulum sheets and mitochondria regulate mitochondrial DNA replication and segregation. iScience 2023, 26, 107180, https://doi.org/10.1016/j.isci.2023.107180.
- 28.Arguello, T.; Peralta, S.; Antonicka, H.; et al. ATAD3A has a scaffolding role regulating mitochondria inner membrane structure and protein assembly. Cell Rep. 2021, 37, 110139–110139, https://doi.org/10.1016/j.celrep.2021.110139.
- 29.Wu, W.; Lin, C.; Wu, K.; et al. FUNDC 1 regulates mitochondrial dynamics at the ER –mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384, https://doi.org/10.15252/embj.201593102.
- 30.Harada, T.; Sada, R.; Osugi, Y.; et al. Palmitoylated CKAP4 regulates mitochondrial functions through an interaction with VDAC2 at ER-mitochondria contact sites. J. Cell Sci. 2020, 133, jcs249045, https://doi.org/10.1242/jcs.249045.
- 31.Rühmkorf, A.; Harbauer, A.B. Role of Mitochondria–ER Contact Sites in Mitophagy. Biomolecules 2023, 13, 1198, https://doi.org/10.3390/biom13081198.
- 32.Zhang, L.; Yan, F.; Li, L.; et al. New focuses on roles of communications between endoplasmic reticulum and mitochondria in identification of biomarkers and targets. Clin. Transl. Med. 2021, 11, e626, https://doi.org/10.1002/ctm2.626.
- 33.Lim, D.; Dematteis, G.; Tapella, L.; et al. Ca2+ handling at the mitochondria-ER contact sites in neurodegeneration. Cell Calcium 2021, 98, 102453, https://doi.org/10.1016/j.ceca.2021.102453.
- 34.Pichla, M.; Sneyers, F.; Stopa, K.B.; et al. Dynamic control of mitochondria-associated membranes by kinases and phosphatases in health and disease. Cell. Mol. Life Sci. 2021, 78, 6541–6556, https://doi.org/10.1007/s00018-021-03920-9.
- 35.Morgado-Cáceres, P.; Liabeuf, G.; Calle, X.; et al. The aging of ER-mitochondria communication: A journey from undifferentiated to aged cells. Front Cell Dev Biol, 2022, 10, 946678, https://doi.org/10.3389/fcell.2022.946678
- 36.Bergami, M.; Motori, E. Reweaving the Fabric of Mitochondrial Contact Sites in Astrocytes. Front. Cell Dev. Biol. 2020, 8, 592651, https://doi.org/10.3389/fcell.2020.592651.
- 37.Yu, F.; Courjaret, R.; Assaf, L.; et al. Mitochondria-ER contact sites expand during mitosis. iScience 2024, 27, 109379, https://doi.org/10.1016/j.isci.2024.109379.
- 38.De Strooper, B.; Scorrano, L. Close encounter: mitochondria, endoplasmic reticulum and Alzheimer’s disease. EMBO J. 2012, 31, 4095–4097, https://doi.org/10.1038/emboj.2012.279.
- 39.Li, Z.; Cao, Y.; Pei, H.; et al. The contribution of mitochondria-associated endoplasmic reticulum membranes (MAMs) dysfunction in Alzheimer’s disease and the potential countermeasure. Front. Neurosci. 2023, 17, 1158204, https://doi.org/10.3389/fnins.2023.1158204.
- 40.Wilson, E.L.; Metzakopian, E. ER-mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ 2021, 28, 1804–1821.
- 41.Dentoni, G.; Castro-Aldrete, L.; Naia, L.; et al. The Potential of Small Molecules to Modulate the Mitochondria–Endoplasmic Reticulum Interplay in Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 920228, https://doi.org/10.3389/fcell.2022.920228.
- 42.Jagtap, Y.A.; Kumar, P.; Kinger, S.; et al. Disturb mitochondrial associated proteostasis: Neurodegeneration and imperfect ageing. Front Cell Dev Biol. 2023, 11, 1146564. https://doi.org/10.3389/fcell.2023.1146564
- 43.Area-Gomez, E.; Schon, E.A. Towards a Unitary Hypothesis of Alzheimer’s Disease Pathogenesis. J. Alzheimer’s Dis. 2024, 98, 1243‒1275, https://doi.org/10.3233/jad-231318.
- 44.Xu, L.; Wang, X.; Tong, C. Endoplasmic Reticulum–Mitochondria Contact Sites and Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 428, https://doi.org/10.3389/fcell.2020.00428.
- 45.Han, J.; Park, H.; Maharana, C.; et al. Alzheimer’s disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics 2021, 11, 8855–8873.
- 46.Barodia, S.K.; Prabhakaran, K.; Karunakaran, S.; et al. Editorial: Mitochondria and Endoplasmic Reticulum Dysfunction in Parkinson’s Disease. Front Neurosci, 2019, 13, 1171, https://doi.org/10.3389/fnins.2019.01171.
- 47.Xu, J.; Minobe, E.; Kameyama, M. Ca2+ Dyshomeostasis Links Risk Factors to Neurodegeneration in Parkinson’s Disease. Front. Cell. Neurosci. 2022, 16, 867385, https://doi.org/10.3389/fncel.2022.867385.
- 48.Guillén-Samander, A.; De Camilli, P. Endoplasmic Reticulum Membrane Contact Sites, Lipid Transport, and Neurodegeneration. Cold Spring Harb Perspect Biol. 2023, 15, a041257.
- 49.Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front Cell Dev Biol. 2020, 8, 615461, https://doi.org/10.3389/fcell.2020.615461.
- 50.Maity, S.; Komal, P.; Kumar, V.; et al. Impact of ER Stress and ER-Mitochondrial Crosstalk in Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 780, https://doi.org/10.3390/ijms23020780.
- 51.Johri, A.; Chandra, A.; Connection Lost, MAM: Errors in ER–Mitochondria Connections in Neurodegenerative Diseases. Brain Sci, 2021, 11, 1437, https://doi.org/10.3390/brainsci11111437.
- 52.Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca(2+) efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol Dis. 2020, 136, 104741, https://doi.org/10.1016/j.nbd.2020.104741.
- 53.Bernal, A.F.; Mota, N.; Pamplona, R.; et al. Hakuna MAM-Tata: Investigating the role of mitochondrial-associated membranes in ALS. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2023, 1869, 166716, https://doi.org/10.1016/j.bbadis.2023.166716.
- 54.Martín-Guerrero, S.M.; Markovinovic, A.; Mórotz, G.M.; et al. Targeting ER-Mitochondria Signaling as a Therapeutic Target for Frontotemporal Dementia and Related Amyotrophic Lateral Sclerosis. Front. Cell Dev. Biol. 2022, 10, 915931, https://doi.org/10.3389/fcell.2022.915931.
- 55.Chen, J.; Bassot, A.; Giuliani, F.; et al. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789, https://doi.org/10.3390/cells10071789.
- 56.Hartopp, N.; Markovinovic, A.; Miller, C.C.J.; et al., Insight into endoplasmic reticulum-mitochondria contacts in human amyotrophic lateral sclerosis. Neural. Regen. Res. 2024, 19, 1407‒1408.
- 57.Wilson, E.L.; Yu, Y.; Leal, N.S.; et al. Genome-wide CRISPR/Cas9 screen shows that loss of GET4 increases mitochondria-endoplasmic reticulum contact sites and is neuroprotective. Cell Death Dis. 2024, 15, 1–16, https://doi.org/10.1038/s41419-024-06568-y.
- 58.Yang, S.; Zhou, R.; Zhang, C.; et al. Mitochondria-Associated Endoplasmic Reticulum Membranes in the Pathogenesis of Type 2 Diabetes Mellitus. Front. Cell Dev. Biol. 2020, 8, 571554, https://doi.org/10.3389/fcell.2020.571554.
- 59.Cheng, H.; Gang, X.; He, G.; et al. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. 2020, 11, 592129 https://doi.org/10.3389/fendo.2020.592129.
- 60.Thivolet, C.; Vial, G.; Cassel, R.; et al. Reduction of endoplasmic reticulum- mitochondria interactions in beta cells from patients with type 2 diabetes. PLOS ONE 2017, 12, e0182027–e0182027, https://doi.org/10.1371/journal.pone.0182027.
- 61.Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. 2018, 9, 1–12, https://doi.org/10.1038/s41419-018-0416-1.
- 62.Dingreville, F.; Panthu, B.; Thivolet, C.; et al. Differential Effect of Glucose on ER-Mitochondria Ca(2+) Exchange Participates in Insulin Secretion and Glucotoxicity-Mediated Dysfunction of beta-Cells. Diabetes, 2019, 68, 1778–1794.
- 63.Song, H.; Zhang, X.; Wang, J.; et al. The regulatory role of adipocyte mitochondrial homeostasis in metabolism-related diseases. Front. Physiol. 2023, 14, 1261204, https://doi.org/10.3389/fphys.2023.1261204.
- 64.Xia, W.; Veeragandham, P.; Cao, Y.; et al. Obesity causes mitochondrial fragmentation and dysfunction in white adipocytes due to RalA activation. Nat. Metab. 2024, 6, 273–289, https://doi.org/10.1038/s42255-024-00978-0.
- 65.Beaulant, A.; Dia, M.; Pillot, B.; et al. Endoplasmic reticulum-mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J. Hepatol. 2022, 77, 710–722, https://doi.org/10.1016/j.jhep.2022.03.017.
- 66.de Almeida, M.E.; Ørtenblad, N.; Petersen, M.H.; et al. Acute exercise increases the contact between lipid droplets and mitochondria independently of obesity and type 2 diabetes. J. Physiol, 2023, 601, 1797–1815.
- 67.Wang, J.; He, W.; Tsai, P.-J.; et al. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Heal. Dis. 2020, 19, 1–19, https://doi.org/10.1186/s12944-020-01210-0.
- 68.Barbier-Torres, L.; Fortner, K.A.; Iruzubieta, P.; et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 2020, 11, 1–15, https://doi.org/10.1038/s41467-020-16991-2.
- 69.Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041, https://doi.org/10.1016/j.biopha.2021.112041.
- 70.Jin, C.; Felli, E.; Lange, N.F.; et al. Endoplasmic Reticulum and Mitochondria Contacts Correlate with the Presence and Severity of NASH in Humans. Int. J. Mol. Sci. 2022, 23, 8348, https://doi.org/10.3390/ijms23158348.
- 71.Myint, M.; Oppedisano, F.; De Giorgi, V.; et al. Inflammatory signaling in NASH driven by hepatocyte mitochondrial dysfunctions. J. Transl. Med. 2023, 21, 1–16, https://doi.org/10.1186/s12967-023-04627-0.
- 72.Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895, https://doi.org/10.1016/j.cell.2019.04.010.
- 73.Forte, M.; Schirone, L.; Ameri, P.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2020, 178, 2060–2076, https://doi.org/10.1111/bph.15068.
- 74.Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases. Front. Cell Dev. Biol. 2020, 8, 604240, https://doi.org/10.3389/fcell.2020.604240.
- 75.Li, J.; Zhang, D.; Brundel, B.J.; et al. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells 2019, 8, 1617, https://doi.org/10.3390/cells8121617.
- 76.Moro, L. Mitochondria at the Crossroads of Physiology and Pathology. J. Clin. Med. 2020, 9, 1971, https://doi.org/10.3390/jcm9061971.
- 77.Peoples, J.N.; Saraf, A.; Ghazal, N.; et al. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13, https://doi.org/10.1038/s12276-019-0355-7.
- 78.Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; et al. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658, https://doi.org/10.3390/ijms20071658.
- 79.Peruzzo, R.; Costa, R.; Bachmann, M.; et al. Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis. Cancers 2020, 12, 2574, https://doi.org/10.3390/cancers12092574.
- 80.Bustos, G.; Ahumada-Castro, U.; Silva-Pavez, E.; et al. The ER-mitochondria Ca(2+) signaling in cancer progression: Fueling the monster. Int. Rev. Cell Mol. Biol, 2021, 363, 49–121.
- 81.Reyes-Castellanos, G.; Hadi, N.A.; Carrier, A. Autophagy Contributes to Metabolic Reprogramming and Therapeutic Resistance in Pancreatic Tumors. Cells 2022, 11, 426, https://doi.org/10.3390/cells11030426.
- 82.Themistocleous, S.; Christodoulou, P.; Kyriakou, T.-C.; et al. Key genes expressed in mitochondria-endoplasmic reticulum contact sites in cancer (Review). Oncol. Rep. 2023, 49, 1–14, https://doi.org/10.3892/or.2023.8514.
- 83.Nobili, A.; Krashia, P.; D’Amelio, M. Cisd2: a promising new target in Alzheimer’s disease(dagger). J. Pathol. 2020, 251, 113–116.
- 84.Yeh, C.-H.; Chou, Y.-J.; Kao, C.-H.; et al. Mitochondria and Calcium Homeostasis: Cisd2 as a Big Player in Cardiac Ageing. Int. J. Mol. Sci. 2020, 21, 9238, https://doi.org/10.3390/ijms21239238.
- 85.Li, S.X.; Li, J.; Dong, L.W.; et al. Cytoskeleton-Associated Protein 4, a Promising Biomarker for Tumor Diagnosis and Therapy. Fron. Mole. Biosci., 2021, 7, 552056.
- 86.Simoes, I.C.M.; Morciano, G.; Lebiedzinska-Arciszewska, M.; et al. The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2020, 1866, 165834, https://doi.org/10.1016/j.bbadis.2020.165834.
- 87.Grespi, F.; Vianello, C.; Cagnin, S.; et al. The Interplay of Microtubules with Mitochondria–ER Contact Sites (MERCs) in Glioblastoma. Biomolecules 2022, 12, 567, https://doi.org/10.3390/biom12040567.
- 88.Sammeta, S.S.; Banarase, T.A.; Rahangdale, S.R.; et al. Molecular understanding of ER-MT communication dysfunction during neurodegeneration. Mitochondrion 2023, 72, 59–71, https://doi.org/10.1016/j.mito.2023.07.005.
- 89.Hajimahdi, Z. Small Molecules as Protein-Protein Interaction Inhibitors. Iran, J. Pharm. Res, 2016, 15, 1–2.
- 90.Hartopp, N.; Lau, D.H.W.; Martin-Guerrero, S.M.; et al. Disruption of the VAPB-PTPIP51 ER-mitochondria tethering proteins in post-mortem human amyotrophic lateral sclerosis. Front. Cell Dev. Biol. 2022, 10, 950767, https://doi.org/10.3389/fcell.2022.950767.
- 91.Li, M.; Zhang, Y.; Yu, G.; et al. Mitochondria‐associated endoplasmic reticulum membranes tethering protein VAPB‐PTPIP51 protects against ischemic stroke through inhibiting the activation of autophagy. CNS Neurosci. Ther. 2024, 30, e14707, https://doi.org/10.1111/cns.14707.
- 92.Zhang, C.; Liu, B.; Sheng, J.; et al. Potential targets for the treatment of MI: GRP75-mediated Ca2+ transfer in MAM. Eur. J. Pharmacol. 2024, 971, 176530, https://doi.org/10.1016/j.ejphar.2024.176530.
- 93.Wu, H.; Wang, Y.; Li, W.; et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898, https://doi.org/10.1080/15548627.2019.1596482.
- 94.Zhang, T.; Kho, D.H.; Wang, Y.; et al. Gp78, an E3 Ubiquitin Ligase Acts as a Gatekeeper Suppressing Nonalcoholic Steatohepatitis (NASH) and Liver Cancer. PLOS ONE 2015, 10, e0118448, https://doi.org/10.1371/journal.pone.0118448.
- 95.Zhao, H.; Song, G.; Zhu, H.; et al. Pharmacological Effects of Urolithin A and Its Role in Muscle Health and Performance: Current Knowledge and Prospects. Nutrients 2023, 15, 4441, https://doi.org/10.3390/nu15204441.
- 96.Mazarakis, N.; Snibson, K.; Licciardi, P.V.; et al. The potential use of L-sulforaphane for the treatment of chronic inflammatory diseases: A review of the clinical evidence. Clin. Nutr. 2020, 39, 664–675, https://doi.org/10.1016/j.clnu.2019.03.022.
- 97.Rong, Y.P.; Bultynck, G.; Aromolaran, A.S.; et al., The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. 2009, 106, 14397‒14402.
- 98.Janer, A.; Morris, J.L.; Krols, M.; et al. ESYT1 tethers the ER to mitochondria and is required for mitochondrial lipid and calcium homeostasis. Life Sci. Alliance 2023, 7, e202302335, https://doi.org/10.26508/lsa.202302335.
- 99.Huo, Z.; Gu, J.; He, T. Apelin-13 reduces high glucose-induced mitochondrial dysfunction in cochlear hair cells by inhibiting endoplasmic reticulum stress. Exp. Ther. Med. 2024, 27, 1–8, https://doi.org/10.3892/etm.2024.12515.
- 100.Su, Z.D.Z.; Li, C.Q.; Wang, H.W.; et al. Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1 alleviates atherosclerosis through the modulation of M1 polarization. J. Transl. Med. 2023, 21, 427.
- 101.Li, F.; Aljahdali, I.A.M.; Ling, X. Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein–Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6206, https://doi.org/10.3390/ijms23116206.
- 102.Thai, P.N.; Seidlmayer, L.K.; Miller, C.; et al. Mitochondrial Quality Control in Aging and Heart Failure: Influence of Ketone Bodies and Mitofusin-Stabilizing Peptides. Front. Physiol. 2019, 10, 382, https://doi.org/10.3389/fphys.2019.00382.
- 103.Oh, J.G.; Kim, J.; Jang, S.P.; et al. Decoy peptides targeted to protein phosphatase 1 inhibit dephosphorylation of phospholamban in cardiomyocytes. J. Mol. Cell. Cardiol. 2013, 56, 63–71.
- 104.Peretti, D.; Kim, S.; Tufi, R.; et al. Lipid Transfer Proteins and Membrane Contact Sites in Human Cancer. Front. Cell Dev. Biol. 2020, 7, 371, https://doi.org/10.3389/fcell.2019.00371.
- 105.Xia, Y.; Zhang, Y.; Sun, Y.; et al. CCDC127 regulates lipid droplet homeostasis by enhancing mitochondria-ER contacts. Biochem. Biophys. Res. Commun. 2023, 683, 149116, https://doi.org/10.1016/j.bbrc.2023.10.048.
- 106.Muallem, S.; Chung, W.Y.; Jha, A.; et al. Lipids at membrane contact sites: cell signaling and ion transport. Embo Rep. 2017, 18, 1893–1904, https://doi.org/10.15252/embr.201744331.
- 107.Lin, H.; Guo, X.; Liu, J.; et al. Ethanol‐Induced Hepatic Ferroptosis Is Mediated by PERK‐Dependent MAMs Formation: Preventive Role of Quercetin. Mol. Nutr. Food Res. 2024, 68, e2300343, https://doi.org/10.1002/mnfr.202300343.
- 108.Kerkhofs, M.; Bultynck, G.; Vervliet, T.; et al. Therapeutic implications of novel peptides targeting ER–mitochondria Ca2+-flux systems. Drug Discov. Today 2019, 24, 1092–1103, https://doi.org/10.1016/j.drudis.2019.03.020.
- 109.Gambardella, J.; Morelli, M.B.; Wang, X.; et al. The discovery and development of IP3 receptor modulators: an update. Expert Opin. Drug Discov. 2021, 16, 709–718, https://doi.org/10.1080/17460441.2021.1858792.
- 110.Zakyrjanova, G.F.; Gilmutdinov, A.; Tsentsevitsky, A.N.; et al. Olesoxime, a cholesterol-like neuroprotectant restrains synaptic vesicle exocytosis in the mice motor nerve terminals: Possible role of VDACs. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2020, 1865, 158739, https://doi.org/10.1016/j.bbalip.2020.158739.
- 111.Chen, W.; Shen, Z.; Dong, W.; et al. Polygonatum sibiricum polysaccharide ameliorates skeletal muscle aging via mitochondria-associated membrane-mediated calcium homeostasis regulation. Phytomedicine 2024, 129, 155567.
- 112.Wang, J.; Jiang, J.; Hu, H.; et al. MCU complex: Exploring emerging targets and mechanisms of mitochondrial physiology and pathology. J. Adv. Res. 2024, https://doi.org/10.1016/j.jare.2024.02.013.
- 113.Wang, S.; Hu, B.; Ding, Z.; et al. ATF6 safeguards organelle homeostasis and cellular aging in human mesenchymal stem cells. Cell Discov. 2018, 4, 2, https://doi.org/10.1038/s41421-017-0003-0.
- 114.Huang, J.; Wan, L.; Lu, H.; et al. High expression of active ATF6 aggravates endoplasmic reticulum stress-induced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol. Med. Rep. 2018, 17, 6483–6489, https://doi.org/10.3892/mmr.2018.8658.
- 115.Burkewitz, K.; Feng, G.; Dutta, S.; et al. Atf-6 Regulates Lifespan through ER-Mitochondrial Calcium Homeostasis. Cell Rep. 2020, 32, 108125.
- 116.Rossi, A.; Galla, L.; Gomiero, C.; et al. Calcium Signaling and Mitochondrial Function in Presenilin 2 Knock-Out Mice: Looking for Any Loss-of-Function Phenotype Related to Alzheimer’s Disease. Cells 2021, 10, 204, https://doi.org/10.3390/cells10020204.
- 117.Thoudam, T.; Chanda, D.; Lee, J.Y.; et al. Enhanced Ca2+-channeling complex formation at the ER-mitochondria interface underlies the pathogenesis of alcohol-associated liver disease. Nat. Commun. 2023, 14, 1–18, https://doi.org/10.1038/s41467-023-37214-4.
- 118.Giorgi, C.; Bonora, M.; Sorrentino, G.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. 2015, 112, 1779–1784, https://doi.org/10.1073/pnas.1410723112.
- 119.Naia, L.; Pinho, C.M.; Dentoni, G.; et al. Neuronal cell-based high-throughput screen for enhancers of mitochondrial function reveals luteolin as a modulator of mitochondria-endoplasmic reticulum coupling. BMC Biol. 2021, 19, 1–21, https://doi.org/10.1186/s12915-021-00979-5.
- 120.Ntalouka, F.; Tsirivakou, A. Luteolin: A promising natural agent in management of pain in chronic conditions. Front. Pain Res. 2023, 4, 1114428, https://doi.org/10.3389/fpain.2023.1114428.
- 121.Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 1–25, https://doi.org/10.1038/s41392-023-01547-9.
- 122.Rakhmatullina, D.; Mazina, A.; Ponomareva, A.; et al. Mdivi-1 Induced Mitochondrial Fusion as a Potential Mechanism to Enhance Stress Tolerance in Wheat. Life 2022, 12, 1386, https://doi.org/10.3390/life12091386.
- 123.Muñoz, J.P.; Basei, F.L.; Rojas, M.L.; et al. Mechanisms of Modulation of Mitochondrial Architecture. Biomolecules 2023, 13, 1225, https://doi.org/10.3390/biom13081225.
- 124.Chen, L.; Li, Y.; Zambidis, A.; et al. ATAD3A: A Key Regulator of Mitochondria-Associated Diseases. Int. J. Mol. Sci. 2023, 24, 12511, https://doi.org/10.3390/ijms241512511.
- 125.de Marañón, A.M.; Díaz-Pozo, P.; Canet, F.; et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox. Biol. 2022, 53, 102342.
- 126.Distelmaier, F.; Visch, H.-J.; Smeitink, J.A.M.; et al. The antioxidant Trolox restores mitochondrial membrane potential and Ca2+-stimulated ATP production in human complex I deficiency. J. Mol. Med. 2009, 87, 515–522, https://doi.org/10.1007/s00109-009-0452-5.
- 127.Höing, S.; Yeh, T.-Y.; Baumann, M.; et al. Dynarrestin, a Novel Inhibitor of Cytoplasmic Dynein. Cell Chem. Biol. 2018, 25, 357–369, https://doi.org/10.1016/j.chembiol.2017.12.014.
- 128.Rout, S.K.; Priya, V.; Setia, A.; et al. Mitochondrial targeting theranostic nanomedicine and molecular biomarkers for efficient cancer diagnosis and therapy. Biomed Pharmacother 2022, 153, 113451.
- 129.Bano, I.; Butt, U.D.; Mohsan, S.A.H. New challenges in drug discovery. In Novel Platforms for Drug Delivery Applications. 2023, pp. 619–643. https://doi.org/10.1016/B978-0-323-91376-8.00021-5
- 130.Ezike, T.C.; Okpala, U.S.; Onoja, U.L.; et al. Advances in drug delivery systems, challenges and future directions. Heliyon 2023, 9, e17488.
- 131.Aoyama-Ishiwatari, S.; Hirabayashi, Y. Endoplasmic Reticulum–Mitochondria Contact Sites—Emerging Intracellular Signaling Hubs. Front. Cell Dev. Biol. 2021, 9, 653828, https://doi.org/10.3389/fcell.2021.653828.
- 132.Dhanasekaran, S.; Venugopal, D.; Al-Dayan, N.; et al. Emerging insights into mitochondria-specific targeting and drug delivering strategies: Recent milestones and therapeutic implications. Saudi, J. Biol. Sci. 2020, 27, 3581–3592, https://doi.org/10.1016/j.sjbs.2020.07.030.
- 133.Sulaimon, L.A.; Afolabi, L.O.; Adisa, R.A.; et al. Pharmacological significance of MitoQ in ameliorating mitochondria-related diseases. Adv. Redox Res. 2022, 5, 100037, https://doi.org/10.1016/j.arres.2022.100037.
- 134.Milane, L.S.; Dolare, S.; Ren, G.; et al. Combination Organelle Mitochondrial Endoplasmic Reticulum Therapy (COMET) for Multidrug Resistant Breast Cancer. J. Control. Release 2023, 363, 435–451, https://doi.org/10.1016/j.jconrel.2023.09.023.
- 135.Wang, W.; Zhang, Y.; Wang, Z.; et al. A Native Drug-Free Macromolecular Therapeutic to Trigger Mutual Reinforcing of Endoplasmic Reticulum Stress and Mitochondrial Dysfunction for Cancer Treatment. ACS Nano 2023, 17, 11023–11038, https://doi.org/10.1021/acsnano.3c03450.
- 136.Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244, https://doi.org/10.1016/j.ebiom.2021.103244.

How to Cite
Monaghan, R. M. Emerging and Novel Therapeutic Treatments Targeting Mitochondrial-Endoplasmic Reticulum Contact Sites in Metabolic and Vascular Disorders. International Journal of Drug Discovery and Pharmacology 2024, 3 (2), 100008. https://doi.org/10.53941/ijddp.2024.100008.
RIS
BibTex
Copyright & License

Copyright (c) 2024 by the authors.
This work is licensed under a Creative Commons Attribution 4.0 International License.
Contents
References
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com