
Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Expert review
p21-Activated Kinases Present a New Drug Target for Hypertrophic Cardiomyopathy
Yu He, and Ming Lei *
Department of Pharmacology, University of Oxford, Mansfield Road, Oxford, OX1 3QT, UK
* Correspondence: ming.lei@pharm.ox.ac.uk
Received: 17 February 2023
Accepted: 26 March 2023
Published: 28 September 2023
Abstract: Hypertrophic cardiomyopathy (HCM), primarily involving mutations in sarcomeric proteins, is the most common form of inherited heart disease and a leading cause of sudden death in young adults and athletes. HCM patients present with cardiac hypertrophy, fibrosis, and diastolic dysfunction often in a progressive manner. Despite significant progress made in understanding the molecular genetic basis of HCM, there remains a lack of effective and specific treatment for preventing disease progression in HCM. This article first provides an overview of recent progress in understanding the pathogenic basis of disease progression in HCM, in particular dysfunctional calcium handling, mitochondrial impairment, and endoplasmic reticulum stress. This article then analyses the evidence for critical roles of the multifunctional enzymes P21-activated kinase-1 and 2 (Pak1/2) in the heart and our opinion on their therapeutic value as a promising druggable target in pathological hypertrophy and associated ventricular arrhythmias.
Keywords:
Hypertrophic cardiomyopathy p21-activated kinase 1 and 2 endoplasmic reticulum stress mitochondrial impairment calcium handling1. Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease with a prevalence of 1:250–1:500 and is a leading cause of sudden death in young adults and athletes particularly [1]. Genetic studies indicated that HCM-causing mutations are commonly found in sarcomere genes (e.g., MYH7, MYBPC3, and ACTC1) [2]. Significant progress has been made in identifying HCM disease-causing gene mutations and in developing clinical interventions for symptoms and risk mitigation of sudden death [3]. However, there is still a lack of effective and specific treatments for mitigating disease progression in HCM [4]. This may reflect the major challenge of treating genetic mutations and the resulting complicated pathogenesis of the disease. Furthermore, nearly two third of patients with HCM do not have a known pathogenic sarcomere gene variant, but instead have, primarily polygenic, non-sarcomeric gene contributions [5]. Gene therapies currently are not positioned to target polygenic traits and promising sarcomere-targeting pharmacological interventions may also fail to treat these patients [6]. Thus, there is a need to develop a new therapeutic strategy targting the pathogenesis of disease progression in HCM beyond the canonical sarcomere focus.
2. Recent Developments in Understanding HCM Pathogenesis
The initial discovery of causative sarcomeric mutations originally provided insight into the hereditary nature of HCM. However, the pathogenesis of HCM from a single mutation to comprehensive myocardial remodelling, responsible for the disease onset and progression, has not yet been well elucidated. Existing evidence suggests that cellular and subcellular abnormalities triggered by myofilament Ca2+ hypersensitivity in HCM-affected cardiomyocytes play a key causal role in the pathogenesis of HCM [7–10]. However, a number of previously unrecognised non-myofilament dysfunctional subcellular compartments are increasingly implicated in HCM pathogenesis [10–14]. The major pathogenic contributors, canonical and recently discovered, are discussed below:
2.1. Dysfunctional Calcium Handling Contributes to Pro-hypertrophic Signalling and Contractile Dysfunction
Commonly, HCM-related mutant sarcomere proteins, especially mutations in thin filament proteins, induce a persistent increase in myofilament response to Ca2+ with a hypercontractile phenotype [15]. Aberrant myofilament Ca2+ sensitivity has been regarded as the most important initiator for dysfunctional calcium handling in HCM cardiomyocytes [7]. Schober et al. [7] showed that increased myofilament Ca2+ sensitivity in mutant myocytes isolated from mice expressing HCM-associated troponin T (TnT) mutants (TnT-I79N, TnT-F110I, TnT-R278C) produced an increase in cytosolic Ca2+ binding and end-diastolic (Ca2+ i) during steady-state pacing. These changes were accompanied by an increase in afterdepolarizations and triggered activity, which was likely a result of excessive SR Ca2+ release and cytosolic Ca2+ accumulation. These defects ultimately appear to converge on energy deficiency and altered Ca2+ handling as major common paths leading to the anatomic (hypertrophy, myofiber disarray, and fibrosis) and functional features (pathological signalling and cardiac systolic and diastolic dysfunction) characteristic of HCM [16]. Consistently, the Watkins Group also demonstrated that mutant cardiomyocytes have higher Ca2+ buffering, increased diastolic (Ca2+) and slowed Ca2+ reuptake in guinea pig ventricular myocytes expressing HCM-causing mutant variants of human troponin-T, troponin-I, and α-tropomyosin (R92Q, R145G, and D175N) [8]. These changes are coupled with a significant decrease in basal sarcomere length and slowed relaxation. Furthermore, such altered Ca2+ homeostasis is associated with hypertrophic calcineurin/NFAT and extracellular signal-regulated kinase pathways [8]. Similarly, abnormal Ca2+ handling in induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from HCM patients with diastolic dysfunction has been reported by Wu et al. [17]. The importance of altered Ca2+ homeostasis in disease progression of HCM has been also demonstrated via neonatal gene transfer of Serca2a delaying and/or preventing development of hypertrophy and improving cardiac function in Tm180 HCM mice by Pena et al. [18]. Thus, these findings implicate, primarily diastolic, Ca2+ cycling dysfunction as a causative mechanism in HCM pathogenesis in a conserved manner across species.
2.2. Role of Mitochondrial Impairment in HCM Pathogenesis
The importance of mitochondrial dysfunction in HCM pathogenesis is also supported by a number of studies. Metabolic perturbation has been well-integrated into the discussion of HCM pathogenesis. The major consequences of increased sarcomeric Ca2+ sensitivity are increased cross-bridge turnover and high actin-activated ATPase activity [19]. HCM mutations were therefore predicted to generate higher-energy metabolic costs to produce a given tension (“tension cost”) with consequential metabolic energy stress [11–13]. Earlier work first suggested this perturbation was associated with subcellular abnormalities: mitochondrial ultrastructural abnormalities were found in cardiomyocytes from HCM mice with a mutant myosin heavy chain gene (MyHC) [9] or with a mutant cardiac troponin T (R92Q) gene [10]. Decreases in mitochondrial respiration rate and the activity of NADH-linked electron transport chain complex I were observed in mutant MyHC cardiac mitochondria and such decreases preceded cardiac hemodynamic dysfunction [9]. Such work implicates mitochondrial dysfunction across monogenic HCM animal models. More recently, a study by Ranjbarvaziri et al. [10] demonstrated that perturbed metabolic signalling and mitochondrial dysfunction are common features also in cardiac tissue from patients with HCM. Decreases in oxidative phosphorylation capacity and the activity and expression of several mitochondrial complex components were also observed [10]. Furthermore, disease-associated mitochondrial DNA (mtDNA) variants were detected in 11% of HCM patients in a previous study [20]. We have also recently identified a new monogenic mutation in C1QBP in a 14-year old boy presenting with myocardial hypertrophy, exercise intolerance, ptosis, and increased serum lactate [21]. The 9-year old brother has similar clinical manifestations apart from ptosis. In both patients the symptoms began from infancy [21]. C1QBP is a gene encoding a promiscuous protein involved in many processes, but is important in mitochondrial biogenesis [21]. A recent study identified mitochondrial-related multigene variants in HCM patients without sarcomere gene mutations. Among the 212 patients, although they found pathogenic variants in sarcomere-associated genes were more prevalent in non-apical HCM than apical HCM, MT-RNR2 mutations positively correlated with apical HCM [22]. Thus, there is growing evidence that mitochondrial dysfunction contributes to and may even be a hallmark of HCM pathogenesis across: species, monogenic versus polygenic forms, and sarcomeric versus non-sarcomeric mutations.
2.3. Role of Endoplasmic Reticulum (ER) Stress in HCM Pathogenesis
ER stress is likely another important mechanism that underlies disease progression in HCM. Beyond the role of the ER in protein synthesis and folding, the ER regulates vast transcriptional and translational programmes responsible for protein translocation, calcium homeostasis, and biosynthesis of lipids and steroids [23,24]. ER function is intimately connected with that of the mitochondria through Ca2+ signalling and stress signalling cascades [25]. ER stress has not yet been comprehensively investigated in HCM. Nevertheless, a HCM mouse model has been generated by cardiac-specific overexpression of a constitutively active form of calcineurin A (CNA). Bousette et al. [14] used this ER stress HCM model to demonstrate disease-associated upregulation of prominent ER chaperone proteins involved in ER stress response including Grp78, Grp94, and calreticulin, along with upregulation of other markers/mediators of ER stress, including Pdia1, phosphorylation of eIF2α, and splicing of Xbp1. Our unpublished observations in hearts from HCM mice with cardiac actin 1 mutation (Actc1 E99K) supports the presence of ER stress in HCM hearts including the changes in expression and activity of: protein kinase-like ER kinase (PERK); the ER heat shock protein 70 (Hsp70) family member BiP; and C/EBP homologous protein (Chop) for apoptosis. Thus, ER stress presents a previously poorly characterized pathogenic element in HCM with diverse vital cellular functions likely compromised.
As summarised in Figure 1, maladaptive responses in Ca2+ handling, mitochondrial respiration, and ER stress alongside myofilament Ca2+ hypersensitivity in HCM-affected cardiomyocytes present a less well-characterised pathogenic signature resulting in the disease phenotype of HCM. Targeting this less well-characterised signature may provide a potential new therapeutic direction for designing treatment to mitigate disease progression in HCM.
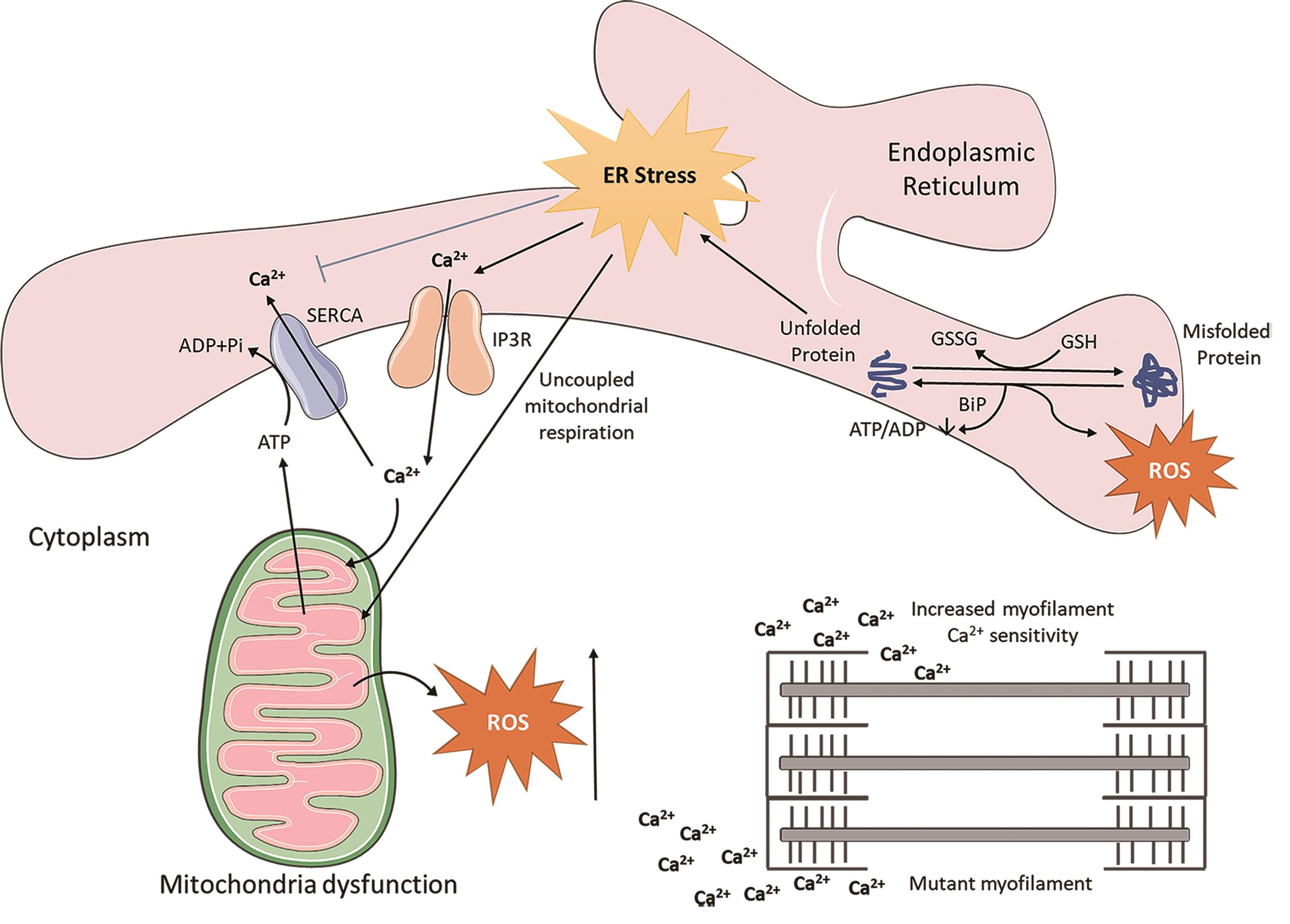
Figure 1. Crosstalk between endoplasmatic reticulum (ER) stress and mitochondrial associated reactive oxygen species (ROS). During the pathological conditions, aggregated proteins result in accumulation of misfolded proteins in the ER lumen, which enhances unfolded protein response induction. ROS are generated in the ER as a part of an oxidative folding process, then ER-induced oxidative stress is further tuned for the generation of mitochondrial ROS through uncoupled mitochondrial respiration. Ca2+ release from the IP3R is stimulated, which increases the production of mitochondrial ROS. Moreover, Ca2+ ions augment the myofilament Ca2+ sensitivity. SERCA, SR Ca2+ -ATPase; IP3R, inositol trisphosphate receptor; GSSG, glutathione disulfide; GSH, glutathione.
3. Current Treatments
Hypertrophic cardiomyopathy often presents in a progression manner and with a high risk in sudden death. The current existing HCM therapies were designed for the purposes of either preventing sudden cardiac death in high‐risk patients via implantation of an ICD [26] and/or improving the symptoms. The current medication consists of beta‐blockers, calcium channel blockers [27], which reduce myocardial energy demand and preventing cytosolic Ca2+ overload and other alternative drugs, such as perhexiline and trimetazidine that affect the myocardial energy metabolism at different levels [28] or inhibit angiotensin (e.g. losartan) [29].
For patients whose symptoms persist, there are also invasive therapies available such as surgical myectomy, alcohol septal ablation, or right ventricular pacing.
A new cardio-selective drug emerged during the past few years, mavacamten, which inhibits myosin binding to actin with good tolerance and has yielded promising results reducing obstruction and improving functionality. Mavacamten was approved in 2022 by the US FDA for the disease specific treatment of obstructive HCM [30,31].
4. PAK1/2 As Therapeutic Targets for the Management of HCM
Group I p21-activated kinases (Paks) are members of the serine/threonine protein kinase family. Paks are encoded by three genes (Pak1‒3) and are involved in the regulation of various biological processes [32]. Pak1 and Pak2 are most homologous isoforms by sharing 91% sequence identity in their kinase domains. Recent studies have shown that Pak1/2 protect the heart from various types of stresses [32]. Activated Pak1/2 participate in the maintenance of normal cellular homeostasis and metabolism, thus enhancing the resilience of cardiomyocytes to adapt to stresses [32]. Over the past decade, a series of studies, including ours, have demonstrated the critical roles of the multifunctional Pak1/2 in the heart and their therapeutic potential as a drug target in pathological hypertrophy and associated ventricular arrhythmias [33–45].
4.1. Pak1 Regulates Contractility, Ca2+ Handling, and Ca2+ Homeostasis
The Solaro Group first demonstrated that activation of Pak1 modulates sarcomeric Ca2+-sensitivity by modification of cTnI protein phosphorylation via PP2A-mediated dephosphorylation [46]. We later identified Pak1 is required to maintain ventricular Ca2+ homeostasis and electrophysiological stability: lack of Pak1 in mice with cardiomyocyte-conditional deletion of Pak1 (Pak1cko) ventricular myocytes led to abnormal Ca2+ homeostasis including increased diastolic (Ca2+)i, altered SR Ca2+ content, and SERCA dysfunction, exacerbated by β-adrenergic stress [41]. Such altered Ca2+ homeostasis is associated with high incidences of electrophysiological instability and ventricular arrhythmias during either acute β-adrenergic or chronic hypertrophic challenge in Pak1cko hearts [41]. Over-expression of constitutively active Pak1 altered Ca2+ transient decay constant (τCa) [47], and promoted anti-adrenergic signalling through attenuation of isoprenaline-induced increases in ICa,L and PLN phosphorylation - both likely through PP2A activation. We also showed that a bioactive peptide (PAP) derived from the Pak1 auto-inhibitory region increases Pak1 activity and counteracts angiotensin II-induced pathological hypertrophy and ventricular arrhythmias [40]. We also demonstrated that FTY720 (Fingolimod), an FDA-approved immuno-modulatory agent, activates Pak1 and reverses existing cardiac hypertrophy and fibrosis caused by pressured overloaded stressor induced by transversal aortic constriction (TAC) [37]. Compared with vehicle-treated TAC hearts, FTY-720 significantly reduced ventricular hypertrophy, ameliorated fibrosis, and improved cardiac performance, further mechanistic studies led to discover that FTY-720 appreciably inhibited nuclear factor of activated T-cells (NFAT) activity [37]. These findings highlight the regulatory role of Pak1, and its potential to reverse cardiac hypertrophy and remodelling.
4.2. Pak2 is Cardioprotective Against ER Stress
The Wang group recently found that Pak2 is abundantly localised in close proximity to the ER membrane and acts as a controller of the IRE1/XBP1-dependent UPR for cardioprotection [44, 45]. Pak2 cardiac-specific deletion mice under tunicamycin-induced ER stress or pressure overload manifested a defective ER stress response, cardiac dysfunction, and profound cardiac cell death. Gene array analysis enabled a detailed mechanistic study, which revealed that Pak2 regulation of protective and appropriate ER stress responses was via the IRE (inositol-requiring enzyme)-1/XBP (X-box–binding protein)-1–dependent pathway. Therapeutically, inducing Pak2 activation by genetic overexpression or viral gene delivery improved ER function and cardiac performance, whilst reducing apoptosis and protecting the heart from failure [44]. Thus, these findings implicate Pak2-mediated modulation of ER stress as cardioprotective in cardiac hypertrophy.
We recently examined the key ER stress markers alongside Pak1/2 expression and phosphorylation in 4- and 7-week-old WT and Actc1E99K HCM mice (reflecting the early stages of HCM). As shown in Figure 2, the expression and phosphorylation of key molecules in 3 branches of ER responses including Atf4, Ire1/Xbp1, and Atf6 in HCM hearts are all down regulated, indicating the ER function in HCM is impaired. Pak1/2 expression is increased in Actc1E99K HCM hearts at both 4 and 7 weeks. However, HCM mouse Pak1/2 phosphorylation appears to be suppressed relative to its protein-level expression. Thus phospho-Pak1 decreases between week 4 and 7, and phospho-Pak2 does not change, both despite the increased available pool of total Pak1/2. Thus, our pilot indicates a disease progression-dependent correlation in alteration of Pak1/2 signalling and ER function. Such preliminary data imply the importance of impaired ER function in the pathogenesis of HCM disease progression, which mandates further detailed interrogation.
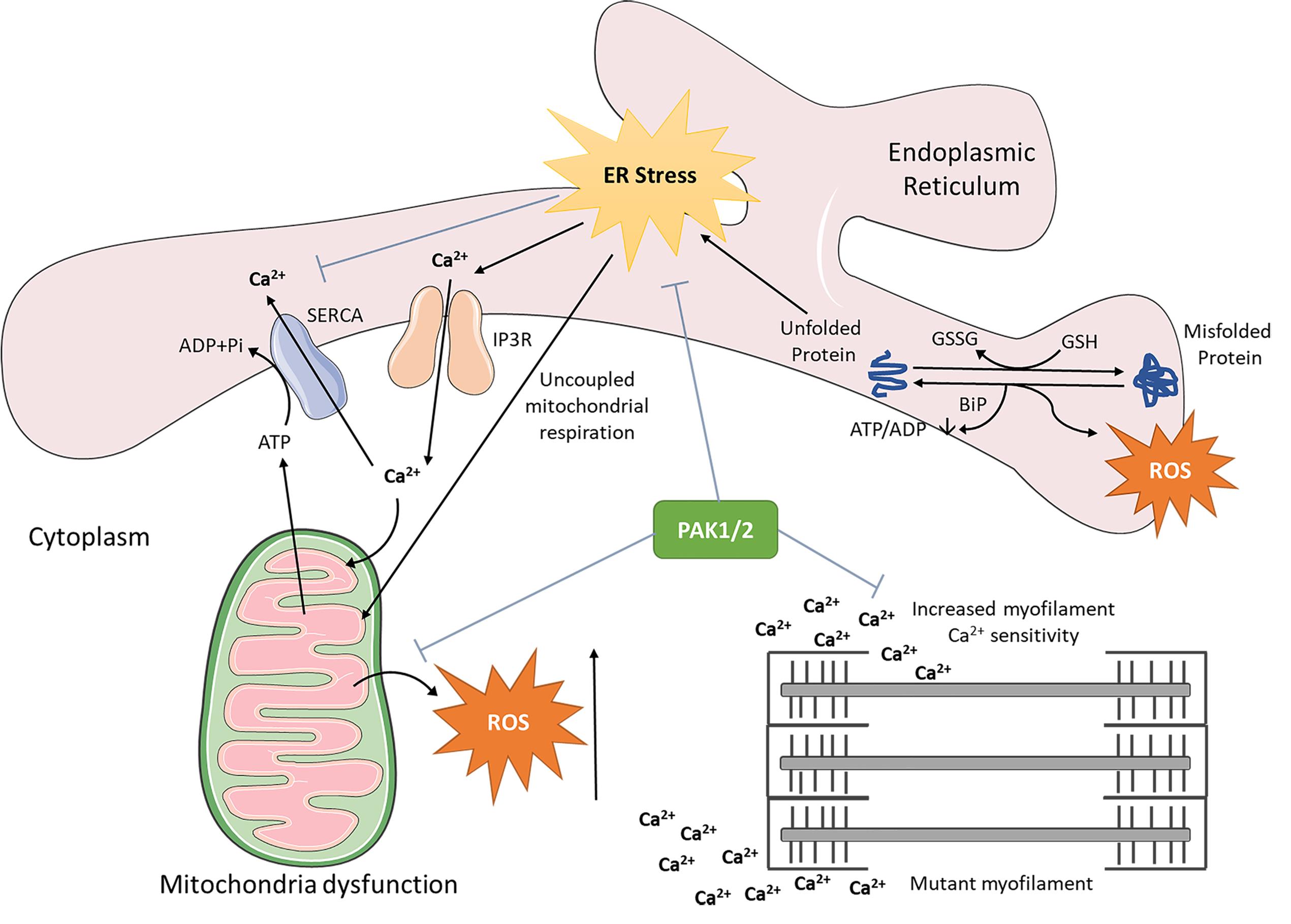
Figure 2. Schematic diagram of Pak1/2 cardiac protective effect against deleterious ER stress and mitochondria dysfunction. PAK, p21 activated kinase.
4.3. The Therapeutic Effect of PAK1 Pharmacological Activation by a Small-molecule in a Mouse HCM Model
Recently, Ryba et al. [48] treated mice, with an HCM-associated mutation in tropomyosin (Tm-E180G) and control wild type littermates, with FTY720 or vehicle for 6 weeks [48]. Compared with vehicle-treated mice, FTY720-treated Tm-E180G mice had a significant reduction in left atrial size and improvement in diastolic function as assessed by echocardiography. However, these anatomical and functional improvements were without any changes in fibrosis. The authors attributed these improvements to a downregulation of S-glutathionylation of cardiac myosin binding protein-C (cMyBP-C) in FTY720-treated Tm-E180G mice and reduction in oxidative stress by downregulation of NADPH oxidases. Their data support the hypothesis that modification of sphingolipid signalling by FTY720 may present a novel therapeutic approach in HCM. An important and novel finding described here is that FTY720 treatment can partially reverse established diastolic dysfunction and atrial remodelling in Tm-E180G mice, by mechanisms likely to be associated with sarcomere activation, reduced expression of NOX2 and NOX4, and decreased S-glutathionylation of cMyBP-C [48].
4.4. PAK1/2 as Therapeutic Target for Designing Novel Antiarrhythmic Drug for Preventing SCD in HCM
Our studies and others have demonstrated the key regulatory and electrophysiological stability in the heart through regulation of calcium handling proteins Ryr2, Serca2a and NCX via either posttranslational and transcriptional mechanisms [41,50], thus providing new insights into atrial Ca2+ homeostasis regulatory mechanisms that have an implication in developing new therapeutic strategies for tachyarrhythmias in HCM and preventing SCD.
5. Conclusions and Future Prospectives
Recent studies indicate that the dysfunctional calcium handling, mitochondrial impairment and endoplasmic reticulum stress are critical pathogenic processes underlying the progression of HCM. Further understanding these processes may provide a potential new therapeutic direction for designing treatment to mitigate or even reverse disease progression in HCM. Pak1/2 activation ameliorates the cardiac remodelling and disease progression of pathological cardiac hypertrophy, HCM, in mouse models, thus providing a new druggable target for the treatment of HCM as proposed in Figure 2. It remains unclear exactly how Pak activation mechanistically leads to these cardioprotective changes, and the extent of the cardioprotection. Future work is required to investigate their downstream signalling pathways, as well as signalling mechanisms specific to PAK1/2 activators in different HCM mouse models, which may pave the way for therapeutic interventions in HCM and other hypertrophic cardiac disease states that currently do not have effective therapies.
Author Contributions: Conceptualization: M.L, draft article: M.L.; H.Y.; A.G.R.; revise and editing: M.L.; H.Y.; A.G.R. have contributed substantially to the work for producing the paper.
Funding: This research was funded by British Heart Foundation (BHF) (BHF Centre for Research Excellence (CRE) at Oxford, PG/14/80/31106, PG/16/67/32340, PG/12/21/29473, PG/21/10512. FS/PhD/20/29653.
Acknowledgments: The authors thank Alexander Grassam-Rowe for his vauable suggestions for improving the manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
References
- Maron B.J.; Desai M.Y.; NishimuraR.A.; et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol., 2022, 79(4): 372-389. DOI: https://doi.org/10.1016/j.jacc.2021.12.002
- Walsh R.; ThomsonK.L.; WareJ.S.; et al. Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med., 2017, 19(2): 192-203. DOI: https://doi.org/10.1038/gim.2016.90
- O'Mahony C.; Akhtar M.M.; Anastasiou Z.; et al. Effectiveness of the 2014 European Society of Cardiology guideline on sudden cardiac death in hypertrophic cardiomyopathy: a systematic review and meta-analysis. Heart, 2019, 105(8): 623-631.
- Prondzynski M.; Mearini G.; Carrier L. Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflügers Archi., 2019, 471(5): 807-815. DOI: https://doi.org/10.1007/s00424-018-2173-5
- Harper A.R.; Goel A.; Grace C.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet., 2021, 53(2): 135-142. DOI: https://doi.org/10.1038/s41588-020-00764-0
- Watkins H. Time to think differently about sarcomere-negative hypertrophic cardiomyopathy. Circulation, 2021, 143(25): 2415-2417. DOI: https://doi.org/10.1161/CIRCULATIONAHA.121.053527
- Schober T.; Huke S.; Venkataraman R.; et al. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ. Res., 2012, 111(2): 170-179. DOI: https://doi.org/10.1161/CIRCRESAHA.112.270041
- Robinson P.; Liu X.; Sparrow A.; et al. Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J. Biol. Chem., 2018, 293(27): 10487-10499. DOI: https://doi.org/10.1074/jbc.RA118.002081
- Lucas D.T.; Aryal P.; Szweda L.I.; et al. Alterations in mitochondrial function in a mouse model of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol., 2003, 284(2): H575-H583. DOI: https://doi.org/10.1152/ajpheart.00619.2002
- Ranjbarvaziri S.; Kooiker B.; Ellenberger M.; et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation, 2021, 144(21): 1714-1731. DOI: https://doi.org/10.1161/CIRCULATIONAHA.121.053575
- Ashrafian H.; Redwood C.; Blair E.; et al. Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet., 2003, 19(5): 263-268. DOI: https://doi.org/10.1016/S0168-9525(03)00081-7
- Sweeney H.L.; Feng H.S.; Yang Z.; et al. Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: insights into disease pathogenesis and troponin function. Proc. Natl. Acad. Sci., 1998, 95(24): 14406-14410. DOI: https://doi.org/10.1073/pnas.95.24.14406
- Frey N.; Brixius K.; Schwinger R.H.G.; et al. Alterations of tension-dependent ATP utilization in a transgenic rat model of hypertrophic cardiomyopathy. J. Biol. Chem., 2006, 281(40): 29575-29582. DOI: https://doi.org/10.1074/jbc.M507740200
- Bousette N.; Chugh S.; Fong V.; et al. Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by α-crystallin-B. Proc. Natl. Acad. Sci., 2010, 107(43): 18481-18486. DOI: https://doi.org/10.1073/pnas.1013555107
- Langa P.; Wolska B.M.; Solaro R.J. The hippo signaling pathway as a drug target in familial dilated cardiomyopathy. Inter. J. Drug Discov. Pharmacol., 2022, 1(1): 4. DOI: https://doi.org/10.53941/ijddp.v1i1.189
- Ashrafian H.; McKenna M.J.; WatkinsH.; et al. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res., 2011, 109(1): 86-96. DOI: https://doi.org/10.1161/CIRCRESAHA.111.242974
- Wu H.D.; Yang H.X.; RheeJ.W.; et al. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J., 2019, 40(45): 3685-3695. DOI: https://doi.org/10.1093/eurheartj/ehz326
- Peña J.R.; Szkudlarek A.C.; Warren C.M.; et al. Neonatal gene transfer of Serca2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol., 2010, 49(6): 993-1002. DOI: https://doi.org/10.1016/j.yjmcc.2010.09.010
- Belus A.; Piroddi N.; Scellini B.; et al. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol., 2008, 586(15): 3639-3644. DOI: https://doi.org/10.1113/jphysiol.2008.155952
- Christiansen M.; Hagen C.M.; Hedley P.L. Mitochondrial haplogroups are associated with hypertrophic cardiomyopathy in the Indian population. Mitochondrion, 2015, 20: 105-106. DOI: https://doi.org/10.1016/j.mito.2014.07.010
- Zhang Y.M.; Wang J.; Zhou Y.F.; et al. Generation of two induced pluripotent stem cell lines (XACHi0010-A, XACHi0011-A) from a Chinese family with combined oxidative phosphorylation deficiency carrying homozygous and heterozygous C1QBP-L275F mutation. Stem Cell Res., 2020, 47: 101912. DOI: https://doi.org/10.1016/j.scr.2020.101912
- Chung H.; Kim Y.; Cho S.M.; et al. Differential contributions of sarcomere and mitochondria-related multigene variants to the endophenotype of hypertrophic cardiomyopathy. Mitochondrion, 2020, 53: 48-56. DOI: https://doi.org/10.1016/j.mito.2020.04.010
- Schwarz D.S.; Blower M.D. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci., 2016, 73(1): 79-94. DOI: https://doi.org/10.1007/s00018-015-2052-6
- Wang S.Y.; Binder P.; Fang Q.R.; et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br. J. Pharmacol., 2018, 175(8): 1293-1304. DOI: https://doi.org/10.1111/bph.13888
- Lin J.H.; Walter P.; Yen T.S.B. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol.: Mech. Dis., 2008, 3: 399-425. DOI: https://doi.org/10.1146/annurev.pathmechdis.3.121806.151434
- Kaski J.P.; Tomé Esteban M.T.T.; Lowe M.; et al. Outcomes after implantable cardioverter-defibrillator treatment in children with hypertrophic cardiomyopathy. Heart, 2007, 93(3): 372-374. DOI: https://doi.org/10.1136/hrt.2006.094730
- Sherrid M.V. Drug therapy for hypertrophic cardiomypathy: physiology and practice. Curr. Cardiol. Rev., 2016, 12(1): 52-65. DOI: https://doi.org/10.2174/1573403X1201160126125403
- Abozguia K.; Elliott P.; McKenna W.; et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation, 2010, 122(16): 1562-1569. DOI: https://doi.org/10.1161/CIRCULATIONAHA.109.934059
- Axelsson A.; Iversen K.; VejlstrupN.; et al. Efficacy and safety of the angiotensin Ⅱ receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol., 2015, 3(2): 123-131. DOI: https://doi.org/10.1016/S2213-8587(14)70241-4
- Heitner S.B.; JacobyD.; LesterS.J.; et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann. Intern. Med., 2019, 170(11): 741-748. DOI: https://doi.org/10.7326/M18-3016
- Keam S.J. Mavacamten: first approval. Drugs, 2022, 82(10): 1127-1135. DOI: https://doi.org/10.1007/s40265-022-01739-7
- Xu H.L.; Wang D.W.; Ramponi C.; et al. The P21-activated kinase 1 and 2 as potential therapeutic targets for the management of cardiovascular disease. Inter. J. Drug Discov. Pharmacol., 2022, 1(1): 5. DOI: https://doi.org/10.53941/ijddp.v1i1.179
- Ke Y.; Lei M.; Collins T.P.; et al. Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ. Res., 2007, 100(9): 1317-1327. DOI: https://doi.org/10.1161/01.RES.0000266742.51389.a4
- Egom E.E.A.; Ke Y.; Musa H.; et al. FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J. Mol. Cell. Cardiol., 2010, 48(2): 406-414. DOI: https://doi.org/10.1016/j.yjmcc.2009.10.009
- Egom E.E.A.; Mohamed T.M.A.; Mamas M.A.; et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am. J. Physiol. Heart Circ. Physiol., 2011, 301(4): H1487-H1495. DOI: https://doi.org/10.1152/ajpheart.01003.2010
- Liu W.; Zi M.; Naumann R.; et al. Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation, 2011, 124(24): 2702-2715. DOI: https://doi.org/10.1161/CIRCULATIONAHA.111.048785
- Liu W.; Zi M.; Tsui H.; et al. A novel immunomodulator, FTY-720 reverses existing cardiac hypertrophy and fibrosis from pressure overload by targeting NFAT (nuclear factor of activated T-cells) signaling and periostin. Circ.: Heart Failure, 2013, 6(4): 833-844. DOI: https://doi.org/10.1161/CIRCHEARTFAILURE.112.000123
- DeSantiago J.; Bare D.; Ke Y.B.; et al. P21-Activated kinase (Pak1) is a negative regulator of ROS generation in ventricular myocytes. Biophys. J., 2013, 104(2 Supplement 1): 614A. DOI: https://doi.org/10.1016/j.bpj.2012.11.3401
- DeSantiago J.; Bare D.J.; XiaoL.; et al. p21-Activated kinase1 (Pak1) is a negative regulator of NADPH-oxidase 2 in ventricular myocytes. J. Mol. Cell. Cardiol., 2014, 67: 77-85. DOI: https://doi.org/10.1016/j.yjmcc.2013.12.017
- Wang R.; Wang Y.W.; Lin W.K.; et al. Inhibition of angiotensin Ⅱ-induced cardiac hypertrophy and associated ventricular arrhythmias by a p21 activated kinase 1 bioactive peptide. PLoS One, 2014, 9(7): e101974. DOI: https://doi.org/10.1371/journal.pone.0101974
- Wang Y.W.; TsuiH.; KeY.B.; et al. Pak1 is required to maintain ventricular Ca2+ homeostasis and electrophysiological stability through SERCA2a regulation in mice. Circ.: Arrhythmia Electrophysiol., 2013, 6(4): 833-844. DOI: https://doi.org/10.1161/CIRCEP.113.001198
- Tsui H.; Zi M.; Wang S.Y.; et al. Smad3 couples Pak1 with the antihypertrophic pathway through the E3 ubiquitin ligase, Fbxo32. Hypertension, 2015, 66(6): 1176-1183. DOI: https://doi.org/10.1161/HYPERTENSIONAHA.115.06068
- Yang B.B.; JiangQ.; HeS.C.; et al. Ventricular SK2 upregulation following angiotensin Ⅱ challenge: modulation by p21-activated kinase-1. J. Mol. Cell. Cardiol., 2022, 164: 110-125. DOI: https://doi.org/10.1016/j.yjmcc.2021.11.001
- Binder P.; Wang S.Y.; Radu M.; et al. Pak2 as a novel therapeutic target for cardioprotective endoplasmic reticulum stress response. Circ. Res., 2019, 124(5): 696-711. DOI: https://doi.org/10.1161/CIRCRESAHA.118.312829
- Binder P.; Nguyen B.; Collins L.; et al. Pak2 regulation of Nrf2 serves as a novel signaling nexus linking ER stress response and oxidative stress in the heart. Front. Cardiovasc. Med., 2022, 9: 851419. DOI: https://doi.org/10.3389/fcvm.2022.851419
- Ke Y.B.; Wang L.; PyleG.; et al. Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ. Res., 2004, 94(2): 194-200. DOI: https://doi.org/10.1161/01.RES.0000111522.02730.56
- Sheehan K.A.; KeY.B.; WolskaB.M.; et al. Expression of active p21-activated kinase-1 induces Ca2+ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am. J. Physiol.: Cell Physiol., 2009, 296(1): C47-C58. DOI: https://doi.org/10.1152/ajpcell.00012.2008
- Ryba D.M.; WarrenC.M.; KaramC.N.; et al. Sphingosine-1-phosphate receptor modulator, FTY720, improves diastolic dysfunction and partially reverses atrial remodeling in a Tm-E180G mouse model linked to hypertrophic cardiomyopathy. Circ.: Heart Failure, 2019, 12(11): e005835. DOI: https://doi.org/10.1161/CIRCHEARTFAILURE.118.005835
- Ke Y.B.; Wang X.; Jin X.Y.; et al. PAK1 is a novel cardiac protective signaling molecule. Front. Med., 2014, 8(4): 399-403. DOI: https://doi.org/10.1007/s11684-014-0380-9
- Wang Y.W.; Tsui H.; BoltonE.L.; et al. Novel insights into mechanisms for Pak1-mediated regulation of cardiac Ca2+ homeostasis. Front. Physiol., 2015, 6: 76. DOI: https://doi.org/10.3389/fphys.2015.00076


