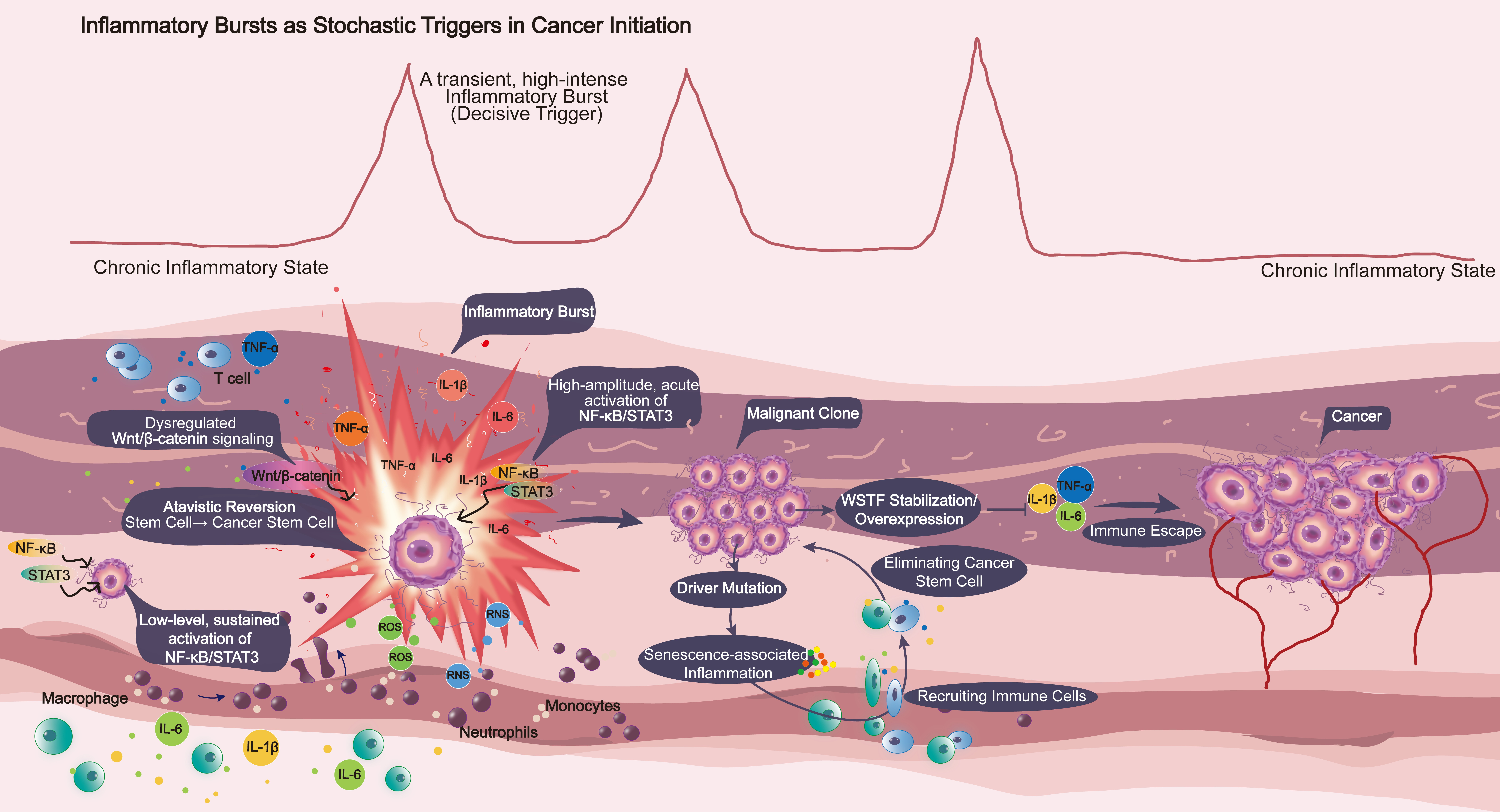
This opinion article proposes the “inflammatory burst” model, positing that intense, transient inflammatory escalations superimposed on chronic inflammation act as decisive stochastic triggers for cancer initiation—complementing the permissive role of somatic mutations. We define inflammatory bursts as high-amplitude, short-lived exacerbations within a chronically inflamed microenvironment, capable of overriding cellular differentiation via sustained nuclear factor kappa B (NF-κB)/signal transducer and activator of transcription 3 (STAT3) activation and dysregulated Wnt/β-catenin signaling, thereby driving an atavistic reversion to a cancer stem cell state. The dual, stage-dependent role of inflammation is illustrated through mechanisms such as Williams syndrome transcription factor (WSTF) nuclear autophagy, which amplifies bursts during initiation but is suppressed to aid immune evasion during progression. This framework shifts the clinical paradigm toward “cancer interception”—a prevention-oriented strategy aimed at selectively dampening pathological inflammatory peaks while preserving immune surveillance, thereby preventing malignant transformation in high-risk individuals.
- Open Access
- Opinion
Inflammatory Bursts as Stochastic Triggers in Cancer Initiation
- Xiuli Zhang,
- Qingshui Wang,
- Yao Lin *
Author Information
Received: 10 Dec 2025 | Revised: 13 Mar 2026 | Accepted: 18 Mar 2026 | Published: 09 Jun 2026
Abstract
Graphical Abstract
Keywords
inflammatory bursts | cancer initiation | chronic inflammation | cancer stem cells | atavistic reversion
References
- 1.
Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. https://doi.org/10.1016/s0092-8674(00)81683-9.
- 2.
Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. https://doi.org/10.1056/NEJMoa1408617.
- 3.
Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. https://doi.org/10.1038/nbt.1554.
- 4.
Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. https://doi.org/10.1016/j.cell.2011.02.013.
- 5.
Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. https://doi.org/10.1093/carcin/bgp127.
- 6.
Shen, G.; Wang, Q.; Li, Z.; Xie, J.; Han, X.; Wei, Z.; Zhang, P.; Zhao, S.; Wang, X.; Huang, X.; et al. Bridging Chronic Inflammation and Digestive Cancer: The Critical Role of Innate Lymphoid Cells in Tumor Microenvironments. Int. J. Biol. Sci. 2024, 20, 4799–4818. https://doi.org/10.7150/ijbs.96338.
- 7.
Aguilar-Cazares, D.; Chavez-Dominguez, R.; Marroquin-Muciño, M.; Perez-Medina, M.; Benito-Lopez, J.J.; Camarena, A.; Rumbo-Nava, U.; Lopez-Gonzalez, J.S. The systemic-level repercussions of cancer-associated inflammation mediators produced in the tumor microenvironment. Front. Endocrinol. 2022, 13, 929572. https://doi.org/10.3389/fendo.2022.929572.
- 8.
Soták, M.; Clark, M.; Suur, B.E.; Börgeson, E. Inflammation and resolution in obesity. Nat. Rev. Endocrinol. 2025, 21, 45–61. https://doi.org/10.1038/s41574-024-01047-y.
- 9.
Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. https://doi.org/10.1016/j.cell.2010.01.025.
- 10.
Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. https://doi.org/10.1016/j.cell.2010.02.029.
- 11.
Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. https://doi.org/10.1016/s0140-6736(00)04046-0.
- 12.
Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. https://doi.org/10.1111/imr.12621.
- 13.
Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. https://doi.org/10.1101/cshperspect.a016295.
- 14.
Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. https://doi.org/10.1038/nri3608.
- 15.
Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. https://doi.org/10.1038/nri3399.
- 16.
Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. https://doi.org/10.1083/jcb.201102095.
- 17.
Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. https://doi.org/10.1038/nri3661.
- 18.
Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. https://doi.org/10.1146/annurev-immunol-030409-101225.
- 19.
Nakagawa, H.; Maeda, S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J. Gastroenterol. 2012, 18, 4071–4081. https://doi.org/10.3748/wjg.v18.i31.4071.
- 20.
Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. https://doi.org/10.1016/j.immuni.2019.06.025.
- 21.
Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. https://doi.org/10.1016/j.cell.2012.12.012.
- 22.
Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. https://doi.org/10.1016/j.stem.2015.02.015.
- 23.
Lineweaver, C.H.; Davies, P.C.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model. Bioessays 2014, 36, 827–835. https://doi.org/10.1002/bies.201400070.
- 24.
Oguma, K.; Oshima, H.; Aoki, M.; Uchio, R.; Naka, K.; Nakamura, S.; Hirao, A.; Saya, H.; Taketo, M.M.; Oshima, M. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. Embo J 2008, 27, 1671–1681. https://doi.org/10.1038/emboj.2008.105.
- 25.
Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. https://doi.org/10.1038/nature03319.
- 26.
Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. https://doi.org/10.1038/ncb2048.
- 27.
Wang, Y.; Eapen, V.V.; Liang, Y.; Kournoutis, A.; Sherman, M.S.; Xu, Y.; Onorati, A.; Li, X.; Zhou, X.; Corey, K.E.; et al. WSTF nuclear autophagy regulates chronic but not acute inflammation. Nature 2025, 644, 780–789. https://doi.org/10.1038/s41586-025-09234-1.
- 28.
Lu, G.; Zhang, S.; Feng, M.; Kim, E.; Cho, D.; Kim, J.H.; Caris, H.; Silberstein, L.; Choi, G.B.; Huh, J.R. In vivo detection of immune responses via cytokine activity labeling. Cell 2026, 189, 939–955.e26. https://doi.org/10.1016/j.cell.2025.12.011.


This work is licensed under a Creative Commons Attribution 4.0 International License.
 Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia
Suite 4002 Level 4, 447 Collins Street, Melbourne, Victoria 3000, Australia General Inquiries: info@sciltp.com
General Inquiries: info@sciltp.com