
Downloads
 Download
Download






This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
The Role of Autophagy in Heart Disease
Osman Mohiuddin 1, Sabu Abraham 1, Hongyuan Zhang 1, and Andrea Ruiz-Velasco 2,*
1 The School of Medical Sciences, The University of Manchester, Oxford Road, Manchester M13 9PT, UK
2 Paris Centre de Recherche Cardiovasculaire, INSERM, 56 Rue Leblanc, 75015 Paris, France
* Correspondence: andrea.ruiz@inserm.fr
Received: 21 June 2024; Revised: 15 July 2024; Accepted: 16 July 2024; Published: 19 November 2024
Abstract: Autophagy is an important biological process occurring in eukaryotic cells. There are different forms of autophagy all of which are specialised for their specific roles. The primary role of autophagy is protein degradation, regulating immune responses and maintaining cellular homeostasis. Due to its complexity, autophagy is modulated by many genes and signalling pathways. Autophagy can be induced via different mechanisms, particularly due to oxidative stress and inflammation resulting in lipid peroxidation products and the generation of cytokines. Dysregulation of autophagy modulation pathways can cause different cardiovascular pathologies ranging from atherosclerosis, cardiac hypertrophy, and failure. Targeting autophagy through therapeutic agents has proven to be fruitful in the treatment of diseases. Potential therapies comprising of Rapamycin, an mTOR inhibitor, and Resveratrol, a polyphenol, have both demonstrated efficacy in reversing cardiac hypertrophy through the modulation of autophagy. However, the challenge lies in translating the studies into real therapies which can be used clinically. It is vital to ensure that the effects of Rapamycin and Resveratrol are safe long-term. Perhaps this can be achieved through further understanding autophagy’s complex interaction with other cellular processes. This literature review will explore the different types of autophagy and their role in normal heart physiology. It also aims to study its relation to the heart diseases mentioned above. Lastly, using autophagy as a tool in disease management will also be discussed.
Keywords:
Autophagy Cardiovascular Pathologies Cellular Homeostasis Oxidative Stress Therapeutic Agents1. Introduction
Autophagy is a phylogenetically preserved degradation and recycling system found within all eukaryote cells [1]. It serves a vital role in cellular homeostasis, protein, and organelle quality control. It is also involved in maintaining cellular balance and regulating immune responses. Due to autophagy being essential for normal physiology, it may not be too surprising that dysregulation in its mechanism can lead to devastating pathologies. Many studies have revealed that dysregulation in autophagy plays a significant role in the onset of disease ranging from the development of tumours, neurological disease, chronic heart disease, and more specifically, cardiovascular disease (CVD).
CVD is one of the major causes of death not only in the UK but also worldwide [2]. Globally, cardiovascular disease remains one of the leading causes of death among the population. It can be split into two main forms: stroke and coronary heart disease (CHD) [3]. In the UK, 27.1% of all deaths recorded in 2019 were identified as being related to cardiovascular disease [4]. Recent studies have highlighted that targeting autophagy can be beneficial in the treatment of CHD. Since then, numerous drugs have emerged as promising candidates in this regard [5].
This literature review aims to provide a comprehensive overview of the role of autophagy in heart disease. Firstly, the mechanism of autophagy will be explored in detail. Next, the role of autophagy in normal heart physiology, particularly in aging, will be discussed. Subsequently, its role in the development of hypertensive heart disease, more specifically, cardiac hypertrophy, failure and atherosclerotic coronary artery disease will be studied. Lastly, this review will also delve into the therapeutic potential of targeting autophagy as a novel approach for heart disease management.
2. Understanding Autophagy
The word autophagy is derived from two Greek words: ‘Autos’ meaning self and ‘Phagomai’ meaning to eat. This ‘self-devouring’ or degradative ability is needed for cell survival. The catabolic process of autophagy can be split into three distinct classes: microautophagy, macroautophagy and chaperone-mediated autophagy (CMA). This is further illustrated in Figure 1.
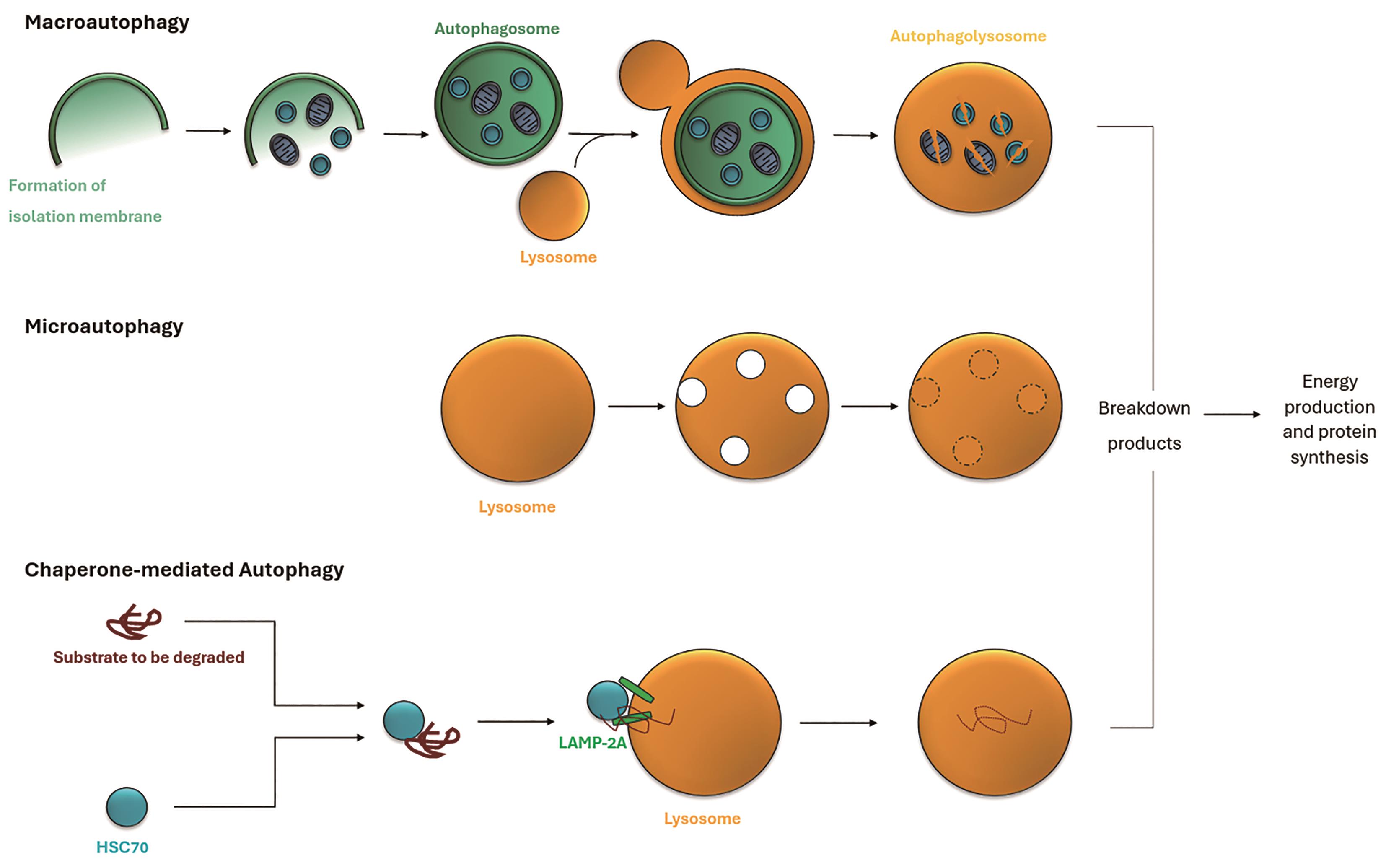
Figure 1. Three types of autophagy: (1) In macroautophagy, an isolation membrane envelops cytoplasmic material and organelles, forming an autophagosome. This fuses with a lysosome, creating an autophagolysosome where the contents are degraded. (2) Microautophagy involves direct engulfment of cytoplasmic parts by the lysosome. (3) Chaperone-mediated autophagy targets specific proteins recognized by HSC70 and translocated into the lysosome via LAMP-2A for degradation. These autophagic processes break down and recycle cytoplasmic material for energy production and protein synthesis. This is an adapted diagram inspired by Mizushima et al. [6], original concept adapted and modified for the purpose of this literature review.
Microautophagy is a process where the cytoplasm itself enters the lysosome and separate vesicles or autophagosomes are not formed [7]. The role of microautophagy in protein degradation remains poorly understood, therefore, it is not extensively covered in this literature review [8].
Chaperone-mediated autophagy (CMA) on the other hand, is much more selective. In CMA, special substrate proteins which need to be degraded are marked with a KFERQ-like sequence [9]. Proteins most commonly marked with a KFERQ-like sequence are either damaged or misfolded and thus need to be destroyed. This KFERQ-like sequence is identified by a cytosolic chaperone protein, namely, Shock Cognate 70 kDa protein (HSC70) [10]. This complex is then translocated, firstly, to the lysosomal membrane. At the membrane the CMA substrate binds to its receptor: lysosomal-associated membrane protein 2A (LAMP2A) [11]. This complex is then ultimately translocated to the lysosome lumen by LAMP2A. Lastly, upon reaching the lumen, the substrate protein is degraded into its basic amino acids by lysosome proteases for recycling. The recycled products can be used for protein synthesis.
This literature review paper will focus on the most common form of autophagy, macroautophagy. Macroautopahgy can be further subcategorised into mitophagy, pexophagy and reticulophagy. Mitophagy is the selective degradation and removal of damaged mitochondria and is essential for maintaining cellular function [12]. Pexophagy on the other hand refers to the selective targeting of peroxisomes. This process regulates the number and the quality of peroxisomes in cells and is crucial for cellular lipid metabolism and detoxification [13]. Lastly, reticulophagy, sometimes also referred to as ER-phagy, is a term for selective degradation of the endoplasmic reticulum (ER). His process helps to maintain ER homeostatsis [14]. From this point on macroautophagy will be referred to as autophagy unless otherwise specified.
2.1. Initiation
The first step of autophagy is initiation. This can be triggered as a response to a number of both intracellular and extracellular stimuli ranging from nutrient deprivation (glucose, amino acids etc.), cellular stress (oxidative stress, DNA damage etc.) and damaged organelles or pathogen infection [15,16]. The key regulator of autophagy is the mammalian target of rapamycin (mTOR) signalling cascade. Normally, target of rapamycin complex 1 (TORC1) is responsible for phosphorylating the autophagy-related unc-51-like kinase (ATG/ULK) complex. This phosphorylation inhibits the synthesis of an autophagosome. Signals from the aforementioned stimuli result in the inactivation of TORC1 inducing autophagy.
2.2. Membrane Nucleation and Expansion
Now that the process of autophagy has been initiated, the next step involves the formation of the phagophore, also known as the isolation membrane. This is a vital step in autophagy as it consists of the assembly of the double membrane autophagosome which engulfs the cytoplasmic material.
In the previous stage of initiation, the ATG/ULK complex was phosphorylated. Research has revealed that this complex is a key player in autophagy [17]. This activated complex then goes on to phosphorylate, and in turn activate, Beclin-1-a member of the phosphatidylinositol 3-kinase (PI3K) complex. PI3K generates phosphatidylinositol 3-phosphate (PI3P) to be formed at the endoplasmic reticulum. PI3P has a vital role in the recruitment of multiple different autophagy related (ATG) proteins.
Before moving onto expansion of the isolation membrane, it is crucial to understand why this is important. Throughout the process of membrane nucleation and expansion, the phagophore is ‘open’. This allows for the entry of cytoplasmic cargo into the developing autophagosome. This differs from the more selective process of CMA where specific proteins are engulfed.
Some examples of recruited ATG proteins and their roles in autophagy nucleation are as follows: ATG18, membrane elongation and recruitment of other ATG proteins. ATG9, a transmembrane protein needed for the delivery of membrane vesicles to the isolation membrane synthesis site [18]. ATG 5, protein which makes up the molecular machinery for the formation of autophagosomes. And lastly, ATG2, which is involved in membrane dynamics and elongation. Once the phagophore reaches sufficient size, ATG2 also initiates phagophore closure [19]. These are just a few autophagy genes; a more comprehensive list can be found in Table 1.
| Gene | Function | Role in Autophagy | Reference |
|---|---|---|---|
| AMPK | AMP-activated protein kinase | Activate | [20] |
| ATG16L | Autophagosome elongation | Activate | [21] |
| ATG2 | Lipidation of ATG8 and autophagosome elongation | Activate | [19] |
| ATG5 | Autophagosome elongation | Activate | [22] |
| ATG7 | E1-like enzyme fort ATG8 conjugation | Activate | [23] |
| ATG8 | Autophagosome formation and elongation | Activate | [24] |
| ATG9 | Membrane protein for autophagosome formation | Activate | [25] |
| BECLIN1 | Autophagosome initiation | Activate | [26] |
| ESCRT | Endosomal sorting complex for pore closure and transport | Activate | [27] |
| mTOR | Component of mTORC1 | Inhibit | [28] |
| p62 | Adaptor protein | Activate | [29] |
| VPS4 | Vacuolar Protein Sorting 4 for autophagosome initiation | Activate | [30] |
2.3. Pore Closure
Once the phagophore has encapsulated the cytoplasmic contents which need to be degraded, the edges move towards each other and ultimately fuse. This membrane fission seals off the phagophore completing the autophagosome. The exact mechanisms in this process are not yet fully understood, however, several key factors can be identified.
Some of the key players in this process, as shown by research, are SNARE proteins which bring the edges of the phagophore towards one another [31]. More importantly, endosomal sorting complexes required for transport (ESCRT) machinery also play a significant role in promoting pore closure in mammals. ESCRT proteins are able to assemble on the autophagosome membrane and though an ATPase, named VPS4, drive membrane sealing [27].
2.4. Degradation
In the last step of autophagy, the previously created autophagosome fuses with a lysosome forming an autophagolysosome [32]. Upon fusion, the cytoplasmic contents are exposed to around 40 different types of hydrolytic digestive enzymes. These enzymes include proteases, nuclease sulfatases and lipases which catalyse the breakdown of different macromolecules [33].
Following degradation, the products generated by the digestive enzymes are released into the cytoplasm for transport to be used in different metabolic processes including protein synthesis and energy production as mentioned earlier in this paper.
3. Autophagy in Normal Heart Physiology
3.1. Exercise and Autophagy
Fascinatingly, many studies have also found a link between exercise and autophagy. It has been reported that mTOR phosphorylation can be inhibited by aerobic exercise [34]. This can directly prevent cardiac aging [35]. The study conducted by Campos et al. highlights this remarkably.
The researchers found that 8 weeks after commencing exercise training, autophagy increased. This resulted in increased mitochondria number, size, and oxidative capacity. Ultimately slowing down cardiac deterioration. Interestingly, the researchers also concluded that when autophagy inhibitors were administered, the protective effect of autophagy and exercise was reversed. The study also found that reduced autophagic flux was found in failing hearts, in turn leading to fragmented mitochondria and impaired oxygen consumption. This is something which will be further explored in detail in the subsequent section [36].
3.2. Autophagy in Aging
Many research studies have shown the link between autophagy and cardiovascular aging. Cardiovascular aging is a process which occurs over a long period of time. There are many hallmarks of cardiac ageing including decreased heart compliance, increase in arterial tree thickness and increased left ventricular afterload [37].
Generally, cardiac aging is characterised by heart hypertrophy and mitochondria dysfunction. As one can imagine, due to autophagy having the role of maintaining cellular and protein homeostasis, it is essential for sustaining cardiac function during aging. This has further been proven with Chang et al.’s roundworm studies [38]. The researchers used fluorescently tagged autophagy-related protein 8 (ATG 8) to visualise autophagy activity in different tissues of wild type worms and two types of long-lived mutants which lack insulin/IGF-1 receptor signalling. Flux assays were also conducted to quantify how many autophagosomes and autolysosomes were present. The findings show an age-dependent increase in autophagosomes and autolysosomes in all wild type worms. This suggests a decrease in autophagic activity as the worms aged.
We know from other studies that impaired autophagy in turn can lead to cardiac dysfunction and ultimately cardiac aging as proven in mice experiments conducted by Taneike et al. [39]. The study demonstrates that impairing autophagy by deletion of autophagy-related protein 5 (ATG 5) in cardiac cells resulted in chamber dilation and cardiac dysfunction with increasing age. Additionally, in cardiac cells with the gene deletion, mitochondria with structural and functional abnormalities were also observed. Furthermore, the accumulation of abnormal mitochondria was linked with increased oxidative stress as levels of heme oxygenase-1 mRNA (HO-1) were found to be elevated. HO-1 is an enzyme which is synthesised in response to oxidative stress and catalyses the breakdown of heme into biliverdin, carbon monoxide and iron [40].
Overall, from analysing these two studies we can conclude that disrupting autophagy by either genetic manipulation (such as ATG 5 ablation) or through age-related decline leads to cardiac dysfunction. This goes to show that continuous autophagy plays a vital role in maintaining cardiac function.
That being said, it is essential to realise that despite both studies providing valuable insight there are a few limitations which come into play. Firstly, the worm and mice models used may not fully recapitulate autophagy in humans. Secondly, there may also be factors other than autophagy which could contribute to the observed age-related dysfunction.
4. Autophagy Triggers
Autophagy in the heart can be triggered due to multiple different factors. A key inducer of autophagy is oxidative stress.
Oxidative stress can result in the formation and accumulation of lipid peroxidation products. An example of this would be the reactive aldehyde 4-hydroxynonenal (4-HNE) [41]. 4-HNE can modify nucleophilic functional groups through the process of covalent adduction. This ultimately can lead to changes in DNA, proteins and lipids. At high levels, 4-HNE can increased cell death through autophagic means [42].
Additionally, autophagy can also be induced and further modulated by cholesterol autoxidation in the presence of oxidative stress. Cholesterol autoxidation is a spontaneous chemical reaction where cholesterol, in the presence of oxygen, undergoes oxidation leading to the formation of products known as oxysterols. A link between cholesterol levels and the initiation autophagy was suggested by a study conducted by Cheng et al. [43]. It was shown that there was an increased level of autophagy related genes following the reduction of cholesterol. The exact process by which lowered cholesterol levels are able to regulate autophagy remains unknown, however [44].
5. Chronic Heart Diseases
As previously mentioned, chronic heart disease is one of the leading causes of death globally. Chronic heart disease refers to long term heart diseases which can impact both the structure and function of the heart leading to severe complications over time. It is a broad term which encompasses many different heart conditions such as coronary artery disease (CAD), heart failure, congenital heart disease, cardiomyopathy, and arrhythmias.
A major risk factor contributing to CHD, as proven by multiple research studies, is hypertension or high blood pressure [45]. Evidence suggests a linear relationship linking the two together [46]. Further research has also shown that in the presence of hypertension, the mortality rate of CHD is 2.3 times greater [47].
Heart disease due to hypertension is referred to as hypertensive heart disease (HHD). It essentially describes changes in heart physiology due to the increased workload resulting from chronically elevated blood pressure. One of the key changes associated with this is an increase in left ventricular wall thickness, left ventricular hypertrophy (LVH).
Before delving deeper into HDD, it is crucial to grasp the underlying cause of LVH. According to Laplace’s law, ventricular wall stress is directly proportional to the pressure and diameter of the ventricle [48]. Also, it is inversely proportional to the wall thickness. Therefore, due to the increased pressure on the ventricular walls in high blood pressure, the wall diameter increases.
Cardiac hypertrophy can further be classified into two categories. Hypertrophy due to pressure overload leads to lateral myocyte growth which in turn results in wall thickening with preserved chamber volume. This is named concentric hypertrophy [49]. On the contrary, in hypertrophy due to volume overload, the sarcomeres are added in series which results in wall thickening and increased chamber volume. This is eccentric hypertrophy [50].
Hypertension not only causes heart failure due to the mechanisms mentioned above, but it can also lead to coronary artery disease through the development of atherosclerotic plaque formation [51].
This paper will specifically focus on heart failure due to hypertensive heart disease as well as coronary heart disease. Recent research has identified autophagy as a key player in these diseases.
5.1. Heart Failure
Heart failure (HF) is a condition in which, due to functional and structural deficits in the myocardium, results in impaired blood ejection or impaired ventricular filling [52]. HF can broadly be categorised into two main types based on ejection fraction: heart failure with preserved ejection fraction (HFpEF) and heart failure with reduced ejection fraction (HFrEF) [53].
Heart failure with reduced ejection fraction, also known as systolic heart failure, results from the left ventricle’s inability to contract effectively, leading to a diminished ejection fraction typically below 40% [32].
In contrast, heart failure with preserved ejection fraction, or diastolic heart failure, arises when the left ventricle exhibits thickened walls but a reduced cavity size, resulting in an increased left ventricular mass-to-volume ratio [54].
5.2. Coronary Artery Disease and Atherosclerosis
Coronary artery disease, also known as coronary heart disease, is a cardiovascular disorder which occurs due to obstruction of the coronary arteries. This obstruction arises primarily due to atherosclerosis which is triggered often due to factors such as high blood pressure but can also be triggered due to injury and damage to the coronary arteries.
Atherosclerosis is a chronic condition characterised by accumulation of plaques found on the walls of arteries. Briefly, disruption of the arterial wall results in lipoprotein droplets to accumulate in the coronary vessels [55]. Water insoluble lipids which generally circulate in the blood stream then attach to these lipoproteins forming apolipoproteins. Apolipoproteins can pass through the vessel endothelium and upon oxidation attract immune cells such as macrophages [56]. This process leads to foamy cell production and ultimately fatty streak lesions. Lesions then attract smooth muscle cells which initiate production and deposition of collogen and proteoglycans which causes progression into fibrous plaque [57].
Factors other than high blood pressure can also contribute to the initiation and progression of atherosclerosis some of which being, high cholesterol levels, smoking, aging and diabetes mellitus [58,59].
6. Autophagy in Chronic Heart Diseases
Many studies have revealed the link between autophagy and hypertensive heart disease. This review will primarily focus on atherosclerosis and the end point of all heart diseases, heart failure.
6.1. Autophagy in Cardiac Hypertrophy and Heart Failure
Autophagy in cardiac hypertrophy has been suggested by many researchers as a double-edged sword. In some cases, dysregulation and an increase in autophagy has promoted cardiac hypertrophy [60]. This increase in autophagy can be triggered by multiple stimuli as presented in Figure 2.
Weng et al. conducted a study demonstrating this effect in mice with transverse aortic constriction (TAC) to induce heart pressure overload. Some mice were administered the renin inhibitor Aliskiren (ALK) which lowers blood pressure and is used to treat hypertension. Rest of the mice received an autophagy inhibitor [61]. The researchers found that TAC induced significant cardiac hypertrophy in mice. Additionally, TAC induced autophagic responses in the heart. This was through increased expression of ATG 5, ATG 16 and BECLIN-1.
Additionally, other studies have shown the role of autophagy in cardiac hypertrophy through the use of miRNAs. In mice with TAC, the expression of miR-30a was shown to be reduced suggesting that it may act as a negative regulator of cardiac hypertrophy. In the study by Yin et al., when a miR-30a inhibitor was used, it resulted in an increase in autophagy and an increase in cardiac hypertrophy markers [62].
The impact of increased autophagy on heart failure was observed in an ultrastructural analysis of patient samples by Fidzianska et al. The examination demonstrated changes in the cardiomyocyte structure due to the presence of large autophagic vacuoles filled with glycogen granules, mitochondria, and cytoplasmic remnants [63]. The vacuoles not only show increased autophagic activity in the cardiomyocytes but also indicate cell stress and damage. This reinforces the involvement of autophagy in the progression of cardiovascular diseases. In this case, the progression from hypertrophy to heart failure [64].
However, every coin has two faces. Several other studies have shown that in some cases, high levels of autophagy may ameliorate heart hypertrophy.
An example of this would be the study conducted by Liu et al. studying the role of Regulated in Development and DNA Damage 1 (REDD1) [65]. REDD1 is a stress responsive protein and is upregulated due to increase cellular stress or DNA damage. In the presence of cardiac hypertrophy, it has been noted that REDD1 is able to inhibit cardiomyocyte growth by increasing autophagy. Furthermore, when REDD1 expression is knocked down, autophagy in cardiomyocytes is reduced.
REDD1 can exert its effects by its regulation of the mTOR pathway. Interestingly, the effect of REDD1 knock out can be counteracted by administration of Rapamycin. This is something which will be explored later in this paper.
Studies show that administration of AMP-activated protein kinase (AMPK) activators such as Metformin increase levels of autophagy in cardiomyocytes and subsequently block hypertrophy [66]. This was observed in in vitro studies and Li et al. suggest this increase in autophagy is triggered by inhibition of the mTOR pathway. The AMPK pathway is also something which will be studied in the subsequent section.
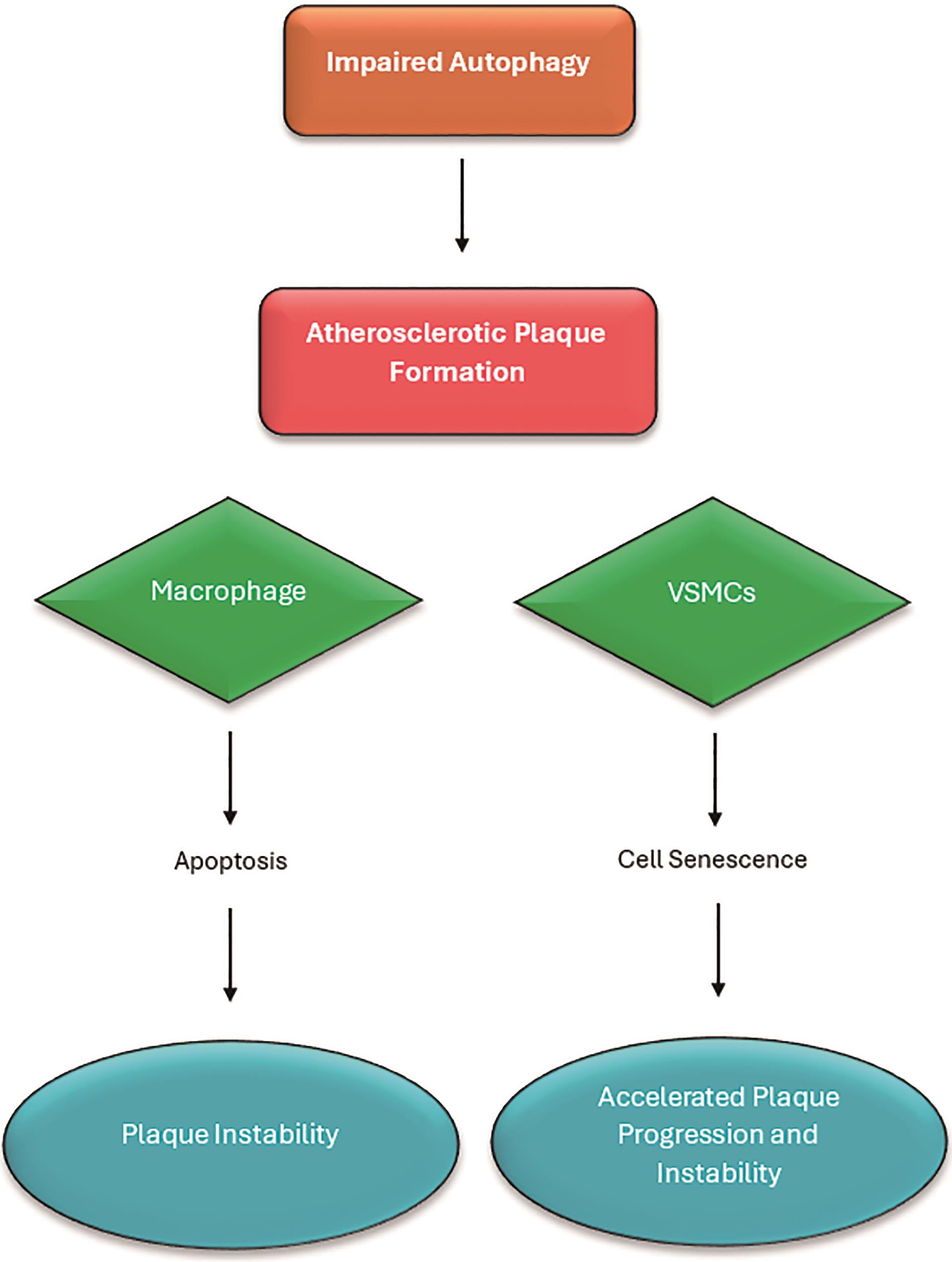
Figure 2. Flowchart explaining the effect of impaired autophagy on different cells which are present in blood vessels. Impaired autophagy in macrophages leads to unstable plaque formation due to apoptosis, in VSMCs cellular senescence results in accelerated play progression and plaque instability. Adapted diagram inspired by Poznyak et al. [67] Original concept adapted and modified for the purpose of this literature review.
6.2. Autophagy in Atherosclerosis
Autophagy resulting in atherosclerotic plaque can be impaired or disrupted in different cell types as illustrated in Figure 3 [67].
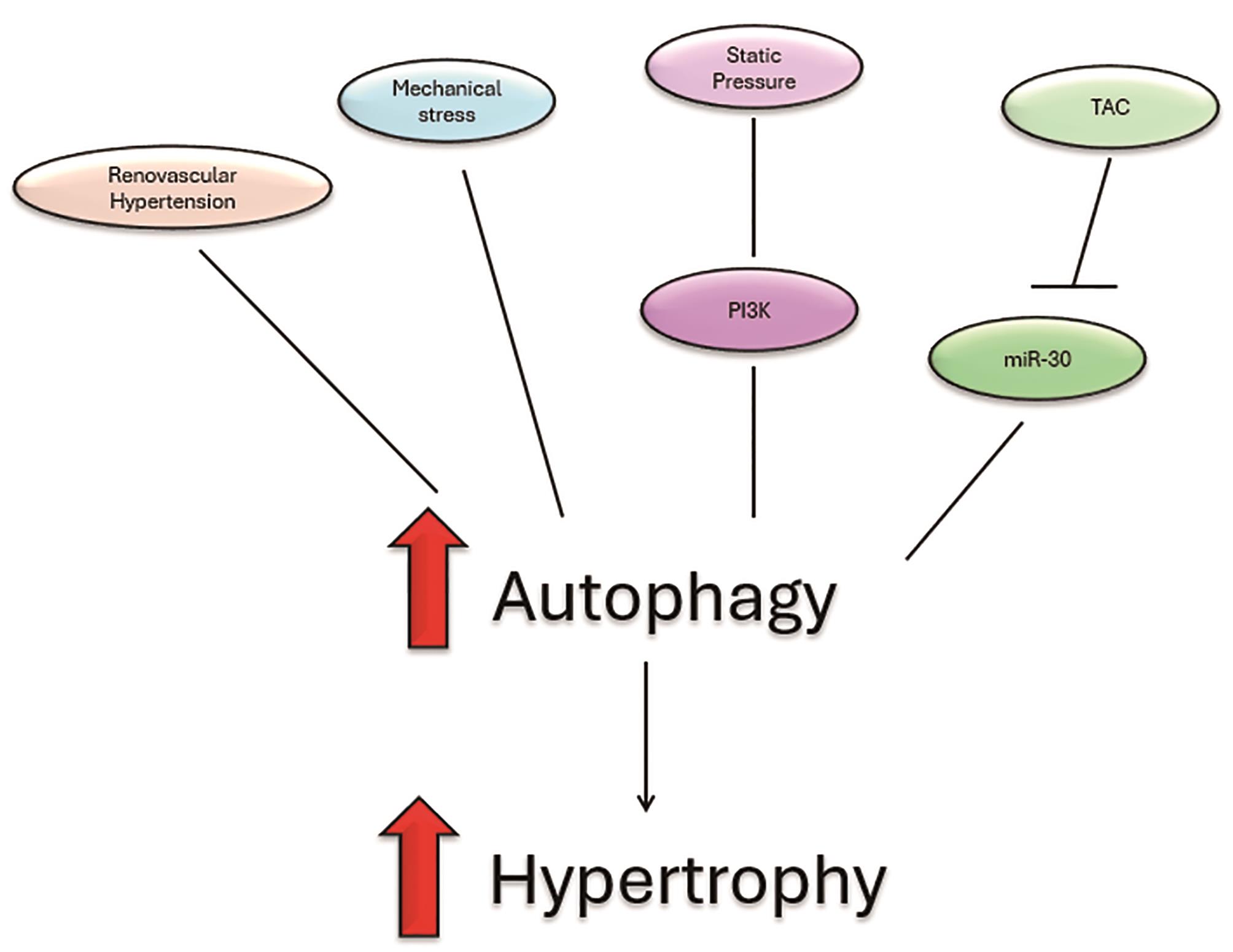
Figure 3. Heart hypertrophy can be induced through factors which increase autophagy. These factors include increased renovascular hypertension, mechanical stress, static pressure though P13K pathway and lastly, transverse aortic constriction which reduced expression of miR-30a. Adapted diagram inspired by Li et al. original concept adapted and modified for the purpose of this literature review.
6.2.1. Autophagy Markers in Macrophages
Analysis in mouse and human tissue by Sergin et al. has revealed that when macrophages are exposed to ‘atherogenic lipids’, an increase of p62 and autophagy-related protein 5 (ATG 5) in both mouse and human tissue samples [68].
Atherogenic lipids specifically refer to low-density lipoprotein (LDL) cholesterol, which, when oxidised, can play a significant role in driving the progression of plaque formation within blood vessels [69]. P62 is a protein found in many different cellular processes but in the context of autophagy, serves as an adapter protein which helps clear damaged cellular components though the process of autophagy [70], while ATG 5, as mentioned earlier, is a protein involved in the formation of autophagosomes.
The research suggests that a deficiency in p62 can lead to the accumulation of protein aggregates and thus exacerbate the progression of atherosclerosis by disrupting autophagy. Furthermore, an impairment in ATG 5 in macrophages in turn leads to an impairment in autophagy due to its protective role of maintaining cellular homeostasis.
Additionally, oxidized LDL (oxLDL) as well as its compounds, 7-ketocholesterol and 7β-hydroxycholesterol can both induce autophagy in vascular wall cells [71]. They are able to induce a special form of cell death termed oxiapoptophagy which is associated with oxidative stress, apoptosis and autophagy [72]. Oxiapoptophagy is especially relevant in atherosclerosis treatment as it highlights core signalling pathways which could be targeted for therapies. An interesting study conducted by Ouyang et al. highlights pretreatment with antioxidant acetylcysteine (NAC). The study suggested that NAC could decrease the production of NOX4 (responsible for NADPH oxidase 4 synthesis), as well as ameliorating expression of autophagy-related factors and decreasing the production of apoptotic proteins [73]. Moreover, a different study also shows NAC induced prevention of atherosclerosis though the reduction of reactive oxidative species formation, oxLDL and inflamitory cytokines [74]. Therefore, it can be concluded that dysfunction in autophagy, as evidenced by alterations in autophagic markers in macrophages, is implicated in the pathogenesis of atherosclerosis [68].
Sergin et al.’s study provides great insight into the relationship between autophagy and atherosclerosis progression, particularly emphasising the significance of role p62. This may partly be due to its well-established role in autophagy. However, the study also prompts further inquiry into the molecular pathways underlying ATG 5 deficiency. Identifying these pathways could unveil novel therapeutic targets for the treatment of atherosclerosis. Thus, this research underscores the importance of ongoing investigation in this field to advance our understanding and therapeutic strategies for cardiovascular diseases.
6.2.2. Dysfunction of Autophagy in Vascular Smooth Muscle Cells
Another investigation conducted by Nahapetyan et al. reveals that development of atherosclerosis can also be a result of a deletion mutation in the autophagy-related gene 7 (ATG 7) in vascular smooth muscle cells (VSMCs) [75]. VSMCs play a key role in atherosclerosis as they contribute to plaque formation. This is possible as they can change phenotype becoming highly proliferative [76]. ATG 7 is a crucial protein needed for the initiation of autophagy.
The reason ATG 7 is so vital is that it facilitates the conjugation of microtubule-associated protein 1A/1B-light chain 3 (LC3) to the phagophore. Thus, a deficiency in ATG 7 disrupts this initiation process.
VSMCs are important when it comes to the development of atherosclerotic plaques. These plaques can become instable and rupture leading to harmful cardiovascular events. The mice studies show that in VSMCs which lack the ATG 7 gene, the impaired autophagy results in the accumulation of fragmented mitochondria.
The research also shows that the fragmented mitochondria produce less energy and lead to increased oxidative stress within the cell [77]. These effects lead to impaired cellular function. Dysfunctional fragmented mitochondria also release reactive oxidative species which can further exacerbate plaque instability. If the defective mitochondria are not cleared it leads to VSMC apoptosis and makes atherosclerotic plaques more vulnerable to rupture.
In addition, there is evidence to suggest that reduced autophagy in endothelial cells leads to accelerated plaque formation also [78]. This is due to similar mechanisms as dysregulated VSMCs lead to increased inflammation and oxidative stress.
The study by Nahapetyan et al. offers interesting links between ATG 7 deficiency and development of atherosclerosis. As mentioned above, we know the link between dysregulated macrophages and autophagy. Perhaps it may be insightful to consider how VSMC’s, endothelial cells and macrophages interact together and the complex interplay between their respective process in the regulation of autophagy. Further research in this field could pave the way for therapeutics which target molecular mechanisms which modulate these interactions.
7. Autophagy Signalling Pathways in Heart Disease
There are many signalling pathways which modulate autophagy. Some key players are mammalian target of Rapamycin (mTOR) and AMP-activated protein kinase (AMPK) [79].
7.1. AMP-Activated Protein Kinase
AMPK is a key regulator of cell metabolism which is activated due to decreased cellular ATP levels or increased cell demand. It is also able to modulate autophagy as shown in Figure 4 though phosphorylation of TSC2 and RAPTOR which inhibits the mTORC1 complex [80]. Other than acting on the mTOR pathway, AMPK also activates ULK1 and BECLIN1 for autophagy initiation [81].
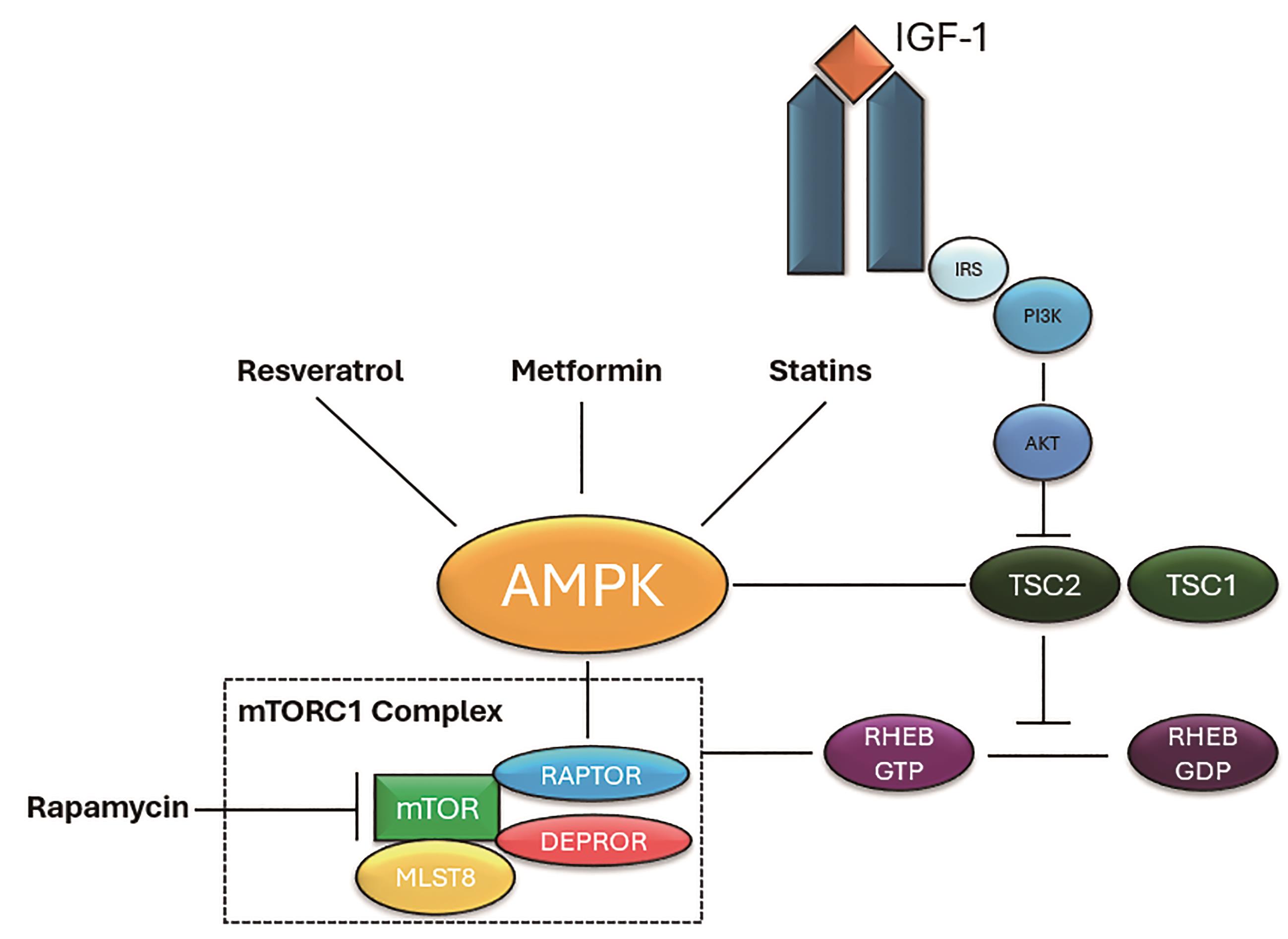
Figure 4. Growth factors such as IGF-1 can activate the PI3L/AKT pathway. Phosphorylation of TSC2 by AKT leads to conversion of RHEB-GDP into its active form, RHEB-GTP, ultimately resulting in the activation of mTORC1. AMPK can be activated by Resveratrol, Metformin, and statins. It can then inactivate mTORC1 by phosphorylating TSC2 upstream or RAPTOR downstream. Adapted diagram inspired by Orogo et al. original concept adapted and modified for the purpose of this literature review.
7.2. mTOR
The primary role of mTOR is integration of signals from growth factors to promote cell growth and inhibit autophagy [82]. It is a kinase which is a part of the mTORC1 complex along with Proline-Rich Akt Substrate of 40 kDa (PRAS40), DEP Domain-Containing mTOR-Interacting Protein (DEPTOR), Regulatory-Associated Protein of mTOR (RAPTOR) and Mammalian Lethal with SEC13 Protein 8 (MLST8).
Mouse model studies have shown that deletion of mTOR in adult heart results in increased apoptosis and autophagy and thus, reduced cell survival [28]. The study goes to show that mosaic deletion of mTOR during early heart development led to apoptosis of cardiomyocytes. Furthermore, when 50% of the cells lacked mTOR, the embryonic heart could not sustain the circulatory requirements and lead to embryonic death. This cements the essential role of mTOR in the survival and proliferation of cardiomyocytes. Disruption of this signalling pathway can lead to cardiac defects such as hypertrophic cardiomyopathy and heart failure [83]. The mTOR pathway regulation and inhibition can be visualised in Figure 4.
8. Therapeutic Potential of Targeting Autophagy in Heart Disease.
As discussed throughout this paper, dysregulation of autophagy can have severe detrimental effects on cardiac function. Therefore, pharmacological modulation of autophagy can be a beneficial approach in the treatment of autophagy related cardiac diseases. Studies have shown multiple drugs and agents which are able to either activate or inhibit autophagy to improve cardiac function.
8.1. Discovery and Development of Autophagy Modulating Drugs
Novel autophagy targets can be identified at a fast rate due to advancements in high-throughput screening (HTS) methods. HTS is a discovery process which allows for the automatic testing of a vast array of chemical and biological compounds for a particular target. Many autophagy modulators have been identified through HTS as highlighted by Deitersen et al. for Arzanol [84].
Another novel drug discovery method being used by scientists is computational modelling and virtual screening [85]. Large libraries of chemical compounds can be screened and certain compounds with the highest probability of binding to a specific target can be identified. Computational modelling can also incorporate machine learning and artificial intelligence to identify patterns in specific datasets which may not be apparent through traditional methods. Computational analysis was used in the study of ULK1 and ULK2 by Dementer et al. to uncover the function of those autophagy proteins [86].
8.1.1. Rapamycin, mTOR Modulation
Rapamycin, also known as Sirolimus, is a naturally occurring compound which can be isolated from Streptomyces hygroscopicus [87]. It is a macrolide compound often used in anticancer therapies and to prevent organ transplant rejection [88,89]. Rapamycin and its derivatives, as shown in Figure 4, are activators of autophagy due to their ability to inhibit mTOR. Studies have shown that Rapamycin is able to protect from cardiac hypertrophy and improve cardiac function.
McMullen et al. found that administration of rapamycin to mice with heart hypertrophy not only led to significant changes in gene expression, but it also regressed both compensated and decompensated heart hypertrophy. Rapamycin reversed the decrease in Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 2a (SERCA2a) gene expression [90]. SERCA2a is important for heart contraction and relaxation as it is involved in the transport of calcium ions. In heart hypertrophy, SERCA2a expression is decreased resulting in the impairment of heart contraction [91]. The authors suggest that Rapamycin may have a therapeutic role for improving cardiac function and regressing cardiac hypertrophy. The study employed a well-designed system to investigate the effect of Rapamycin on mouse models however, despite the findings being promising, more studies need to be conducted to evaluate the effectiveness of rapamycin in humans as well as determining long tern effects and impact on mortality.
A different study conducted by Das et al. demonstrated that Rapamycin treatment on mice with type 2 diabetes prevented cardiac dysfunction [92]. Its cardioprotective effects are achieved, as explained earlier, through mTOR inhibition without interfering with other pathways such as AKT phosphorylation. The study goes on to explain that the cardioprotective effects of rapamycin may be due to its ability to act on many different cellular processes involved in glucose metabolism, antioxidant defence and contractile function.
8.1.2. Resveratrol, AMPK Modulation
Another promising agent in the treatment of cardiac hypertrophy is Resveratrol [93]. It is a naturally occurring polyphenol found in a range of plant species [94]. It is particularly found in red grapes and berries and known to have antibacterial and antifungal properties. Studies have suggested it also acts as a powerful regulator of autophagy [95]. In a study by Chan et al. the potential of the polyphenol to mitigate cardiac hypertrophy was investigated [93]. Both in vitro and in vivo models were employed to investigate its effects on hypertrophy. It was found that treatment with Resveratrol inhibited the growth of cardiomyocytes and reduced the size of the heart in the mice models. Resveratrol was found to be able to activate AMPK and inhibit AKT signalling in cardiomyocytes, as shown in Figure 4. This AMPK activation was associated with promoting energy balance and the inhibition of AKT suppressed the cardiac hypertrophy in cardiomyocytes.
Overall, Chan et al.’s study highlights the anti-hypertrophic effects of Resveratrol in both inhibiting excessive growth in cardiomyocytes as well as balancing cell homeostasis. However, it is important to note that despite its powerful regulative effects, Resveratrol is very rapidly metabolised by the body [96]. Studies showing that both oral and intravenous administration of the drug in humans was quickly metabolised within two hours. And other studies showing that Resveratrol has poor intestinal uptake [97]. Perhaps other analogues of Resveratrol may be more effective in this regard.
8.1.3. PI3K Inhibitors
Phosphoinositide 3-kinases (PI3Ks) are a family of lipid kinases which regulate different intracellular signalling events in the body. The pathways which are regulated by PI3K are essential when it comes to maintaining cellular homeostasis. There are three different classes of PI3Ks. Irregular activity of PI3K has been shown in many different diseases including heart failure. One member of the PI3K family is noted to be of interest, VPS34. This is due to it having an essential role in autophagy. Studies have shown PI3K plays a pivotal role in the transition of cardiac hypertrophy into heart failure due to prolonged activation of autophagy [98]. The mice study conducted by Yu et al. showed that Class III PI3K VPS34 was upregulated in hypertrophic hearts from the age of 3 weeks to 4 weeks. It was also to be noted that the anti-apoptotic factor BCL-2 was decreased by 58.4% when compared to age-matched wild type hearts. The researcher’s data shows that low levels of BCL-2 promote the interaction between VPS34 and Beclin-1, this in turn results in the activation of Class III PI3K, which goes on to stimulate increased autophagic activity in already hypertrophic hearts. Therefore, inhibitors of PI3K, particularly Class III inhibitors, have emerged as promising agents in treatment.
8.2. Therapeutic Challenges of Autophagy Modulation Drugs
As the molecular mechanisms of autophagy are being studied further, multiple different avenues of drug discovery and development targeting this pathway have emerged. Even within the last decade, there have been nearly hundreds of autophagy-related compounds which have both pre-clinical and clinical interest. However, despite all the fascinating breakthroughs in autophagy modulation, there are many problems which need to be addressed [99]. For instance, rapamycin as mentioned earlier, not only targets autophagy, but also targets a wide range of pathways and thus cannot be classified as a selective therapeutic. This lack of specificity may result in off-target effects and a limited efficacy when presented in the clinical setting. Despite McMullen et al.’s study showing promising results, translating drugs into clinical practice after evaluating their safety, efficacy, and long-term effects in human patients can be quite challenging [100]. On the contrary, even more selective targets mentioned in this paper such as PI3K inhibitors need much more refinement before they can even be evaluated and considered for clinical trials [101].
9. Conclusion and Perspective
The link between autophagy and heart disease has emerged to be a promising field in the research of therapeutics. This literature review has explored autophagy in detail, ranging from its different forms, its mechanism, and its intricate role in maintaining homeostasis in the body.
This review has explored a range of cardiovascular diseases and how autophagy can act as a double-edged sword in the onset and progression of these diseases. Namely, dysregulation of autophagy contributing to cardiac hypertrophy, heart failure and atherosclerosis. Lastly, this review has also shed light on autophagy modulation as a potential target in the treatment of hypertensive related heart conditions. This has been possible by analysing preclinical studies which modulate autophagy signalling pathways via Rapamycin and its derivatives as well as Resveratrol. Both are shown to prevent and regress cardiac hypertrophy.
The progress achieved through multiple clinical trials in the field has proven to be remarkable, however, a few challenges lie ahead. As with all clinical trials, it is important to translate the results into effective therapies. Furthermore, as mentioned throughout this paper, the long-term effect and safety of these therapies needs to be evaluated. As autophagy is such a complex process involving many different signalling pathways, it can be difficult to isolate autophagy without altering cell dynamics. As a result, further research needs to be conducted to unravel the crosstalk of autophagy signalling pathways with other cellular processes to observe how potential therapeutics may impact this.
In essence, autophagy plays a pivotal role in the development of cardiovascular diseases and multiple trials and studies have shown considerable progress in targeting autophagy for treatment. Nonetheless, continued effort is needed to fully harness autophagy modulation in order to ensure effective management of heart disease.
Author Contributions: O.M.: conceptualization, writing—original draft preparation, writing—reviewing and editing and visualization. Minor formatting assistance was provided by H.Z.
Funding: This research received no external funding
Institutional Review Board Statement: Not applicable
Informed Consent Statement: Not applicable
Data Availability Statement: Not applicable
Conflicts of Interest: The authors declare no conflict of interest.

References
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3. https://doi.org/10.1002/PATH.2697.
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, 108863. https://doi.org/10.15252/EMBJ.2021108863.
- Price, A.E. Heart Disease and Work. Heart 2004, 90, 1077–1084. https://doi.org/10.1136/HRT.2003.029298.
- Cheema, K.M.; Dicks, E.; Pearson, J.; et al. Long-Term Trends in the Epidemiology of Cardiovascular Diseases in the UK: Insights from the British Heart Foundation Statistical Compendium. Cardiovasc. Res. 2022, 118, 2267–2280. https://doi.org/10.1093/cvr/cvac053.
- Sciarretta, S.; Zhai, P.; Shao, D.; et al. Rheb Is a Critical Regulator of Autophagy during Myocardial Ischemia: Pathophysiological Implications in Obesity and Metabolic Syndrome. Circulation 2012, 125, 1134–1146. https://doi.org/10.1161/CIRCULATIONAHA.111.078212/-/DC1.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. https://doi.org/10.1016/j.cell.2011.10.026.
- Marzella, L.; Ahlberg, J.; Glaumann H. Autophagy, Heterophagy, Microautophagy and Crinophagy as the Means for Intracellular Degradation. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol.1981, 36, 219–234. https://doi.org/10.1007/BF02912068.
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-Known Self-Eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. https://doi.org/10.1007/S00018-011-0865-5/METRICS.
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; et al. Proteome-Wide Analysis of Chaperone-Mediated Autophagy Targeting Motifs. PLoS Biol. 2019, 17. https://doi.org/10.1371/JOURNAL.PBIO.3000301.
- Losmanová T.; Janser, F.A.; Humbert, M.; et al. Chaperone-Mediated Autophagy Markers LAMP2A and HSC70 Are Independent Adverse Prognostic Markers in Primary Resected Squamous Cell Carcinomas of the Lung. Oxid. Med. Cell. Longev. 2020, 2020. https://doi.org/10.1155/2020/8506572.
- Rout, A.K.; Strub, M.P.; Piszczek, G.; et al. Structure of Transmembrane Domain of Lysosome-Associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-Mediated Autophagy. J. Biol. Chem. 2014, 289, 35111–35123. https://doi.org/10.1074/JBC.M114.609446.
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, Pathophysiological Roles, and Analysis. Biol. Chem.2012, 393, 547. https://doi.org/10.1515/HSZ-2012-0119.
- Cho, D.H.; Kim, Y.S.; Jo, D.S.; et al. Pexophagy: Molecular Mechanisms and Implications for Health and Diseases. Mol. Cells 2018, 41, 55. https://doi.org/10.14348/MOLCELLS.2018.2245.
- Cebollero, E.; Reggiori, F.; Kraft C. Reticulophagy and Ribophagy: Regulated Degradation of Protein Production Factories. Int. J. Cell Biol. 2012, 2012, 182834. https://doi.org/10.1155/2012/182834.
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67. https://doi.org/10.1146/ANNUREV-GENET-102808-114910.
- Gross, A.S.; Graef M. Mechanisms of Autophagy in Metabolic Stress Response. J. Mol. Biol. 2020, 432, 28–52. https://doi.org/10.1016/J.JMB.2019.09.005.
- Stjepanovic, G.; Davies, C.W.; Stanley, R.E.; et al. Assembly and Dynamics of the Autophagy-Initiating Atg1 Complex. Proc. Natl. Acad. Sci. USA 2014, 111, 12793–12798. https://doi.org/10.1073/PNAS.1407214111.
- Suzuki, S.W.; Yamamoto, H.; Oikawa, Y.; et al. Atg13 HORMA Domain Recruits Atg9 Vesicles during Autophagosome Formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3350–3355. https://doi.org/10.1073/PNAS.1421092112.
- Velikkakath, A.K.G.; Nishimura, T.; Oita, E.; et al. Mammalian Atg2 Proteins Are Essential for Autophagosome Formation and Important for Regulation of Size and Distribution of Lipid Droplets. Mol. Biol. Cell 2012, 23, 896–909. https://doi.org/10.1091/MBC.E11-09-0785.
- Itakura, E.; Kishi-Itakura, C.; Mizushima N. The Hairpin-Type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. https://doi.org/10.1016/J.CELL.2012.11.001.
- Vietri, M.; Radulovic, M.; Stenmark H. The Many Functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. https://doi.org/10.1038/S41580-019-0177-4.
- MacGurn, J.A. Garbage on, Garbage off: New Insights into Plasma Membrane Protein Quality Control. Curr. Opin. Cell Biol. 2014, 29, 92–98. https://doi.org/10.1016/J.CEB.2014.05.001.
- Rubinsztein, D.C.; Codogno, P.; Levine B.Autophagy Modulation as a Potential Therapeutic Target for Diverse Diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. https://doi.org/10.1038/NRD3802.
- Tamboli, I.Y.; Tien, N.T.; Walter J.The Autophagy Initiating Kinase ULK1 Is Regulated via Opposing Phosphorylation by AMPK and MTOR. Autophagy 2011, 7, 643–644. https://doi.org/10.4161/AUTO.7.6.15123.
- Saitoh, T.; Fujita, N.; Jang, M.H.; et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature 2008, 456, 264–268. https://doi.org/10.1038/NATURE07383.
- Mizushima, N.; Yoshimori, T.; Ohsumi Y.The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. https://doi.org/10.1146/ANNUREV-CELLBIO-092910-154005.
- Komatsu, M.; Tanida, I.; Ueno, T.; et al. The C-Terminal Region of an Apg7p/Cvt2p Is Required for Homodimerization and Is Essential for Its E1 Activity and E1-E2 Complex Formation. J. Biol. Chem. 2001, 276, 9846–9854. https://doi.org/10.1074/JBC.M007737200.
- Wesch, N.; Kirkin, V.; Rogov, V.V.Atg8-Family Proteins-Structural Features and Molecular Interactions in Autophagy and Beyond. Cells 2020, 9. https://doi.org/10.3390/CELLS9092008.
- He, C.; Baba, M.; Cao, Y.; et al. Self-Interaction Is Critical for Atg9 Transport and Function at the Phagophore Assembly Site during Autophagy. Mol. Biol. Cell 2008, 19, 5506. https://doi.org/10.1091/MBC.E08-05-0544.
- Yue, Z.; Jin, S.; Yang, C.; et al. Beclin 1, an Autophagy Gene Essential for Early Embryonic Development, Is a Haploinsufficient Tumor Suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077. https://doi.org/10.1073/PNAS.2436255100.
- Zhu, Y.; Pires, K.M.P.; Whitehead, K.J.; et al. Mechanistic Target of Rapamycin (Mtor) Is Essential for Murine Embryonic Heart Development and Growth. PLoS ONE 2013, 8, 54221. https://doi.org/10.1371/JOURNAL.PONE.0054221.
- Moscat, J.; Diaz-Meco, M.T. P62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 2009, 137, 1001. https://doi.org/10.1016/J.CELL.2009.05.023.
- Hanson, P.I.; Roth, R.; Lin, Y.; et al. Plasma Membrane Deformation by Circular Arrays of ESCRT-III Protein Filaments. J. Cell Biol. 2008, 180, 389–402. https://doi.org/10.1083/JCB.200707031.
- He, C.; Bassik, M.C.; Moresi, V.; et al. Exercise–Induced BCL2–Regulated Autophagy Is Required for Muscle Glucose Homeostasis. Nature 2012, 481, 511. https://doi.org/10.1038/NATURE10758.
- Klionsky, D.J.; Saltiel, A.R. Autophagy Works Out. Cell Metab. 2012, 15, 273. https://doi.org/10.1016/J.CMET.2012.02.008.
- Campos, J.C.; Queliconi, B.B.; Bozi, et al. Exercise Reestablishes Autophagic Flux and Mitochondrial Quality Control in Heart Failure. Autophagy 2017, 13, 1304. https://doi.org/10.1080/15548627.2017.1325062.
- Miyamoto, S. Autophagy and Cardiac Aging. Cell Death Differ. 2019, 26, 653. https://doi.org/10.1038/S41418-019-0286-9.
- Dai, D.F.; Chen, T.; Johnson, S.C.; et al. Cardiac Aging: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 16, 1492. https://doi.org/10.1089/ARS.2011.4179.
- Chang, J.T.; Kumsta, C.; Hellman, A.B.; et al. Spatiotemporal Regulation of Autophagy during Caenorhabditis Elegans Aging. Elife 2017, 6, e18459. https://doi.org/10.7554/ELIFE.18459.
- Taneike, M.; Yamaguchi, O.; Nakai, A.; et al. Inhibition of Autophagy in the Heart Induces Age-Related Cardiomyopathy. Autophagy 2010, 6, 600–606. https://doi.org/10.4161/auto.6.5.11947.
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. https://doi.org/10.1146/ANNUREV.PHARMTOX.010909.105600/CITE/REFWORKS.
- Butterfield, D.A.; Lauderback, C.M. Lipid Peroxidation and Protein Oxidation in Alzheimer’s Disease Brain: Potential Causes and Consequences Involving Amyloid β-Peptide-Associated Free Radical Oxidative Stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. https://doi.org/10.1016/S0891-5849(02)00794-3.
- Sharma, S.; Patel, F.; Ara, H.; et al. Rotenone-Induced 4-HNE Aggresome Formation and Degradation in HL-1 Cardiomyocytes: Role of Autophagy Flux. Int. J. Mol. Sci. 2022, 23, 4675. https://doi.org/10.3390/IJMS23094675.
- Cheng, J.; Ohsaki, Y.; Tauchi-Sato, K.; et al. Cholesterol Depletion Induces Autophagy. Biochem. Biophys. Res. Commun. 2006, 351, 246–252. https://doi.org/10.1016/J.BBRC.2006.10.042.
- Seo, Y.K.; Jeon T., Ⅱ; Chong, H.K.; et al. Genome-Wide Localization of SREBP-2 in Hepatic Chromatin Predicts a Role in Autophagy. Cell Metab. 2011, 13, 367–375. https://doi.org/10.1016/J.CMET.2011.03.005.
- Assmann, G.; Schulte H.The Prospective Cardiovascular Münster (PROCAM) Study: Prevalence of Hyperlipidemia in Persons with Hypertension and/or Diabetes Mellitus and the Relationship to Coronary Heart Disease. Am. Heart J.1988, 116, 1713–1724. https://doi.org/10.1016/0002-8703(88)90220-7.
- Ezzati, M.; Lopez, A.D.; Rodgers, A.; et al. Selected Major Risk Factors and Global and Regional Burden of Disease. Lancet 2002, 360, 1347–1360. https://doi.org/10.1016/S0140-6736(02)11403-6.
- Stamler, J.; Stamler, R.; Neaton, J.D. Blood Pressure, Systolic and Diastolic, and Cardiovascular Risks: US Population Data. Arch. Intern. Med. 1993, 153, 598–615. https://doi.org/10.1001/ARCHINTE.1993.00410050036006.
- Burch, G.E.; Ray, C.T.; Cronvich, J.A. Certain Mechanical Peculiarities of the Human Cardiac Pump in Normal and Diseased States. Circulation 1952, 5, 504–513. https://doi.org/10.1161/01.CIR.5.4.504.
- Levy, D.; Garrison, R.J.; Savage, D.D.; et al. Prognostic Implications of Echocardiographically Determined Left Ventricular Mass in the Framingham Heart Study. N. Engl. J. Med. 1990, 322, 1561–1566. https://doi.org/10.1056/NEJM199005313222203.
- Frey, N.; Katus, H.A.; Olson, E.N.; et al. Hypertrophy of the Heart: A New Therapeutic Target? Circulation 2004, 109, 1580–1589. https://doi.org/10.1161/01.CIR.0000120390.68287.BB.
- Weber, T.; Lang, I.; Zweiker, R.; et al. Hypertension and Coronary Artery Disease: Epidemiology, Physiology, Effects of Treatment, and Recommendations: A Joint Scientific Statement from the Austrian Society of Cardiology and the Austrian Society of Hypertension. Wien. Klin. Wochenschr. 2016, 128, 467–479. https://doi.org/10.1007/S00508-016-0998-5/METRICS.
- Ohtani, T.; Mohammed, S.F.; Yamamoto, K.; et al. Diastolic Stiffness as Assessed by Diastolic Wall Strain Is Associated with Adverse Remodelling and Poor Outcomes in Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2012, 33, 1742. https://doi.org/10.1093/EURHEARTJ/EHS135.
- Kitzman, D.W.; Little, W.C.; Brubaker, P.H.; et al. Pathophysiological Characterization of Isolated Diastolic Heart Failure in Comparison to Systolic Heart Failure. JAMA 2002, 288, 2144–2150. https://doi.org/10.1001/JAMA.288.17.2144.
- Tabas, I.; Williams, K.J.; Borén J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis: Update and Therapeutic Implications. Circulation 2007, 116, 1832–1844. https://doi.org/10.1161/CIRCULATIONAHA.106.676890.
- Tabas, I. Macrophage Apoptosis in Atherosclerosis: Consequences on Plaque Progression and the Role of Endoplasmic Reticulum Stress. Antioxid. Redox Signal. 2009, 11, 2333. https://doi.org/10.1089/ARS.2009.2469.
- Padró T.; Peña, E.; García-Arguinzonis, M.; et al. Low-Density Lipoproteins Impair Migration of Human Coronary Vascular Smooth Muscle Cells and Induce Changes in the Proteomic Profile of Myosin Light Chain. Cardiovasc. Res. 2008, 77, 211–220. https://doi.org/10.1093/CVR/CVM045.
- Libby, P.; Buring, J.E.; Badimon, L.; et al. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. https://doi.org/10.1038/s41572-019-0106-z.
- Abdellatif, M.; Ljubojevic-Holzer, S.; Madeo, F.; et al. Autophagy in Cardiovascular Health and Disease. Prog. Mol. Biol. Transl. Sci. 2020, 172, 87–106. https://doi.org/10.1016/BS.PMBTS.2020.04.022.
- Li, L.; Xu, J.; He, L.; et al. The Role of Autophagy in Cardiac Hypertrophy. Acta Biochim. Biophys. Sin. 2016, 48, 491. https://doi.org/10.1093/ABBS/GMW025.
- Weng, L.Q.; Zhang, W.; Ye, Y.; et al. Aliskiren Ameliorates Pressure Overload-Induced Heart Hypertrophy and Fibrosis in Mice. Acta Pharmacol. Sin. 2014, 35, 1005. https://doi.org/10.1038/APS.2014.45.
- Yin, X.; Peng, C.; Ning, W.; et al. MiR-30a Downregulation Aggravates Pressure Overload-Induced Cardiomyocyte Hypertrophy. Mol. Cell. Biochem. 2013, 379, 1–6. https://doi.org/10.1007/S11010-012-1552-Z.
- Fidziańska, A.; Bilińska, Z.T.; Walczak, E.; et al. Autophagy in Transition from Hypertrophic Cardiomyopathy to Heart Failure. J. Electron Microsc. 2010, 59, 181–183. https://doi.org/10.1093/JMICRO/DFP048.
- Rothermel, B.A.; Hill, J.A. Autophagy in Load-Induced Heart Disease. Circ. Res.2008, 103, 1363. https://doi.org/10.1161/CIRCRESAHA.108.186551.
- Liu, C.; Xue, R.; Wu, D.; et al. REDD1 Attenuates Cardiac Hypertrophy via Enhancing Autophagy. Biochem. Biophys. Res. Commun. 2014, 454, 215–220. https://doi.org/10.1016/J.BBRC.2014.10.079.
- Li, Y.; Chen, C.; Yao, F.; et al. AMPK Inhibits Cardiac Hypertrophy by Promoting Autophagy via MTORC1. Arch. Biochem. Biophys. 2014, 558, 79–86. https://doi.org/10.1016/J.ABB.2014.06.023.
- Poznyak, A.V.; Nikiforov, N.G.; Wu W.-K.; et al. Autophagy and Mitophagy as Essential Components of Atherosclerosis. Cells 2021, 10, 443. https://doi.org/10.3390/cells10020443.
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; et al. Inclusion Bodies Enriched for P62 and Polyubiquitinated Proteins in Macrophages Protect against Atherosclerosis. Sci. Signal. 2016, 9, ra2–ra2. https://doi.org/10.1126/SCISIGNAL.AAD5614/SUPPL_FILE/9_RA2_SM.PDF.
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; et al. The Role of Lipids and Lipoproteins in Atherosclerosis. Science 2019, 111, 166–186. doi: 10.1126/science.111.2877.166
- Liu, W.J.; Ye, L.; Huang, W.F.; et al. P62 Links the Autophagy Pathway and the Ubiqutin-Proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. https://doi.org/10.1186/S11658-016-0031-Z/FIGURES/3.
- He, C.; Zhu, H.; Zhang, W.; et al. 7-Ketocholesterol Induces Autophagy in Vascular Smooth Muscle Cells through Nox4 and Atg4B. Am. J. Pathol. 2013, 183, 626–637. https://doi.org/10.1016/J.AJPATH.2013.04.028.
- Nury, T.; Zarrouk, A.; Yammine, A.; et al. Oxiapoptophagy: A Type of Cell Death Induced by Some Oxysterols. Br. J. Pharmacol. 2021, 178, 3115–3123. https://doi.org/10.1111/BPH.15173.
- Ouyang, J.; Xiao, Y.; Ren, Q.; et al. 7-Ketocholesterol Induces Oxiapoptophagy and Inhibits Osteogenic Differentiation in MC3T3-E1 Cells. Cells 2022, 11, 2882. https://doi.org/10.3390/CELLS11182882.
- Xu, Y.; Bu, H.; Jiang, Y.; et al. N‑acetyl Cysteine Prevents Ambient Fine Particulate Matter‑potentiated Atherosclerosis via Inhibition of Reactive Oxygen Species‑induced Oxidized Low Density Lipoprotein Elevation and Decreased Circulating Endothelial Progenitor Cell. Mol. Med. Rep. 2022, 26, 1–9. https://doi.org/10.3892/MMR.2022.12752.
- Nahapetyan, H.; Moulis, M.; Grousset, E.; et al. Altered Mitochondrial Quality Control in Atg7-Deficient VSMCs Promotes Enhanced Apoptosis and Is Linked to Unstable Atherosclerotic Plaque Phenotype. Cell Death Dis. 2019, 10, 119. https://doi.org/10.1038/S41419-019-1400-0.
- Chen, R.; McVey, D.G.; Shen, D.; et al. Phenotypic Switching of Vascular Smooth Muscle Cells in Atherosclerosis. J. Am. Heart Assoc. 2023, 12, 31121. https://doi.org/10.1161/JAHA.123.031121.
- Markin, A.M.; Khotina, V.A.; Zabudskaya, X.G.; et al. Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis. Life 2021, 11, 165. https://doi.org/10.3390/LIFE11020165.
- Osonoi, Y.; Mita, T.; Azuma, K.; et al. Defective Autophagy in Vascular Smooth Muscle Cells Enhances Cell Death and Atherosclerosis. Autophagy 2018, 14, 1991–2006. https://doi.org/10.1080/15548627.2018.1501132.
- Orogo, A.M.; Gustafsson Å.B. Therapeutic Targeting of Autophagy: Potential and Concerns in Treating Cardiovascular Disease. Circ. Res. 2015, 116, 489. https://doi.org/10.1161/CIRCRESAHA.116.303791.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214. https://doi.org/10.1016/J.MOLCEL.2008.03.003.
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; et al. Phosphorylation of ULK1 (HATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456. https://doi.org/10.1126/SCIENCE.1196371.
- Saxton, R.A.; Sabatini, D.M.MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. https://doi.org/10.1016/J.CELL.2017.03.035.
- Zhang, D.; Contu, R.; Latronico, M.V.G.; et al. MTORC1 Regulates Cardiac Function and Myocyte Survival through 4E-BP1 Inhibition in Mice. J. Clin. Invest. 2010, 120, 2805. https://doi.org/10.1172/JCI43008.
- Deitersen, J.; Berning, L.; Stuhldreier, F.; et al. High-Throughput Screening for Natural Compound-Based Autophagy Modulators Reveals Novel Chemotherapeutic Mode of Action for Arzanol. Cell Death Dis. 2021, 12, 560. https://doi.org/10.1038/s41419-021-03830-5.
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; et al. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334. https://doi.org/10.1124/PR.112.007336.
- Demeter, A.; Romero-Mulero, M.C.; Csabai, L.; et al. ULK1 and ULK2 Are Less Redundant than Previously Thought: Computational Analysis Uncovers Distinct Regulation and Functions of These Autophagy Induction Proteins. Sci. Rep. 2020, 10, 10940. https://doi.org/10.1038/S41598-020-67780-2.
- Kim, Y.H.; Park, B.S.; Bhatia, S.K.; et al. Production of Rapamycin in Streptomyces Hygroscopicus from Glycerol-Based Media Optimized by Systemic Methodology. J. Microbiol. Biotechnol. 2014, 24, 1319–1326. https://doi.org/10.4014/JMB.1403.03024.
- Blagosklonny, M.V. Cancer Prevention with Rapamycin. Oncotarget 2023, 14, 342. https://doi.org/10.18632/ONCOTARGET.28410.
- Augustine, J.J.; Bodziak, K.A.; Hricik, D.E.Use of Sirolimus in Solid Organ Transplantation. Drugs 2007, 67, 369–391. https://doi.org/10.2165/00003495-200767030-00004.
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; et al. Inhibition of MTOR Signaling With Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055. https://doi.org/10.1161/01.CIR.0000130641.08705.45.
- Seth, M.; Sumbilla, C.; Mullen, S.P.; et al. Sarco(Endo)Plasmic Reticulum Ca2+ ATPase (SERCA) Gene Silencing and Remodeling of the Ca2+ Signaling Mechanism in Cardiac Myocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 16683–16688. https://doi.org/10.1073/PNAS.0407537101.
- Das, A.; Durrant, D.; Koka, S.; et al. Mammalian Target of Rapamycin (MTOR) Inhibition with Rapamycin Improves Cardiac Function in Type 2 Diabetic Mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145. https://doi.org/10.1074/JBC.M113.521062.
- Chan, A.Y.M.; Dolinsky, V.W.; Soltys, C.L.M.; et al. Resveratrol Inhibits Cardiac Hypertrophy via AMP-Activated Protein Kinase and Akt. J. Biol. Chem. 2008, 283, 24194. https://doi.org/10.1074/JBC.M802869200.
- Salehi, B.; Mishra, A.P.; Nigam, M.; et al. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. https://doi.org/10.3390/BIOMEDICINES6030091.
- Tian, Y.; Song, W.; Li, D.; et al. Resveratrol As A Natural Regulator Of Autophagy For Prevention And Treatment Of Cancer. Onco. Targets. Ther. 2019, 12, 8601. https://doi.org/10.2147/OTT.S213043.
- Szymkowiak, I.; Kucinska, M.; Murias M. Between the Devil and the Deep Blue Sea—Resveratrol, Sulfotransferases and Sulfatases—A Long and Turbulent Journey from Intestinal Absorption to Target Cells. Molecules 2023, 28, 3297. https://doi.org/10.3390/MOLECULES28083297.
- Wang, Y.; Hong, C.; Wu, Z.; et al. Resveratrol in Intestinal Health and Disease: Focusing on Intestinal Barrier. Front. Nutr. 2022, 9, 848400. https://doi.org/10.3389/FNUT.2022.848400.
- Yu, P.; Zhang, Y.; Li, C.; et al. Class III PI3K-Mediated Prolonged Activation of Autophagy Plays a Critical Role in the Transition of Cardiac Hypertrophy to Heart Failure. J. Cell. Mol. Med. 2015, 19, 1710. https://doi.org/10.1111/JCMM.12547.
- Jiang, M.; Wu, W.; Xiong, Z.; et al. Targeting Autophagy Drug Discovery: Targets, Indications and Development Trends. Eur. J. Med. Chem. 2024, 267, 116117. https://doi.org/10.1016/J.EJMECH.2023.116117.
- Sun, D.; Gao, W.; Hu, H.; et al. Why 90% of Clinical Drug Development Fails and How to Improve It?Acta Pharm. Sin. B 2022, 12, 3049. https://doi.org/10.1016/J.APSB.2022.02.002.
- Zachari, M.; Rainard, J.M.; Pandarakalam, G.C.; et al. The Identification and Characterisation of Autophagy Inhibitors from the Published Kinase Inhibitor Sets. Biochem. J. 2020, 477, 801–814. https://doi.org/10.1042/BCJ20190846.


