
Downloads
Download
Additional Files
Download - Supplemengary Material



This work is licensed under a Creative Commons Attribution 4.0 International License.
Article
Mkk7 Protects Against Cardiac Dysfunction in Heart Failure with Preserved Ejection Fraction
Tayyiba Azam 1, * , Hongyuan Zhang 1, Susanne S. Hille 2, Elizabeth J. Cartwright 1, Oliver J. Müller 2, and Xin Wang 1, *
1 Faculty of Biology, Medicine, and Health, University of Manchester, Oxford Road, M13 9PT, Manchester, UK
2 Department of Internal Medicine III, University of Kiel, Germany; German Centre for Cardiovascular Research (DZHK), 24105 Partner Site Hamburg/Kiel/Lübeck, Germany
* Correspondence: xin.wang@manchester.ac.uk (Xin Wang); tayyiba.azam@manchester.ac.uk (Tayyiba Azam)
Received: 12 June 2023
Accepted: 25 September 2023
Published: 18 March 2024
Abstract: Shifts in epidemiological patterns foretell a rapid increase in the number of patients with heart failure (HF) globally, representing a significant health and economic burden. Heart failure with preserved ejection (HFpEF) is now considered the prevailing subtype of HF, with no effective treatment available to combat this syndrome. Previous studies have highlighted the cardioprotective role of MKK7 during cardiac pathology, however, no extensive research has been performed to examine MKK7 in the context of HFpEF. This study aimed to address this shortcoming by using adeno-associated virus (AAV) 9 to overexpress MKK7 in the two-hit clinically relevant HFpEF mouse model. We report that cardiomyocyte-specific overexpression of MKK7 improved the HFpEF phenotype in mice, by impeding cardiac diastolic dysfunction and myocardial fibrosis. Mechanistically, it was found that MKK7 ameliorated ER stress by maintaining IRE1-XBP1 signalling and blunted CHOP increase in the myocardium. To summarise, MKK7 overexpression holds the ability to protect the myocardium from HFpEF associated pathologies.
Keywords:
HFpEF ER Stress IRE1 XBP11. Introduction
Heart failure with preserved ejection fraction (HFpEF) is a complex clinical syndrome that is characterised by increased myocardial stiffness and reduced ventricular compliance limiting the ability of the heart to maintain cardiac output. Consequently, the patient suffers from symptoms such as exercise intolerance, fluid retention, fatigue and dyspnoea [1]. HFpEF is a multifactorial disorder that is regarded as the cardiac manifestation of systemic metabolic disturbance because it usually concomitantly occurs with other metabolic diseases including, diabetes, hypertension, obesity and ageing. An increase in the incidence of these comorbidities is contributing to the growing prevalence of HFpEF [2].
Another factor significantly contributing to the rising rates of HFpEF, is the lack of treatment options available to manage the disorder. The effectiveness of treatments available to manage HFrEF including, beta-blockers, angiotensin receptor blockers (ARBs) and angiotensin-converting enzyme (ACE) inhibitors, have not shown consistent effectiveness in improving disease outcome in patients suffering from HFpEF. This discrepancy can mainly be attributed to differences in the underpinning mechanisms between HFpEF and HFrEF [3]. Currently, the treatment of HFpEF focuses on addressing the underlying diseases contributing to the development and progression of HFpEF and the management of the associated symptoms in order to improve the overall quality of life. Scientists are now trying the identify novel treatment options to limit the progression and development of HFpEF [4].
The clinical syndrome of HFpEF affects millions of people worldwide, however, despite the magnitude of the problem, the underlying pathogenic processes of the disease are largely unknown. Endoplasmic reticulum (ER) disruption has been associated with a plethora of cardiovascular diseases and recently has been shown to underpin HFpEF progression [5,6]. Clear evidence points to a link between the accumulation of misfolded proteins and aberrant ER stress, causing protein toxicity in cardiomyocytes. Cardiomyocytes possess the ability to alleviate ER stress by activation of the unfolded protein response (UPR) pathway. During acute stress, this pathway primarily acts to enhance protein folding capacity, increase protein degradation and decrease protein synthesis. However, severe or prolonged stress overwhelms the UPR machinery, resulting in apoptosis [7]. The UPR consists of three main signalling pathways, that are activated by a transmembrane protein sensor located on the ER membrane; Protein-kinase RNA-like kinase (PERK), Inositol-requiring enzyme 1 (AzamIRE1) and Activating Transcription Factor 6 (ATF6). Together, these branches work in a coordinated manner to initially mitigate ER stress and restore protein homeostasis [8].
Mitogen activated kinase kinase 7 (MKK7) is a stress-activated kinase, that following extracellular stimuli such as hormones, mitogens, cytokines, growth factors, and stress, is activated to regulate a plethora of cellular processes via a complex interconnected signalling cascade [9]. These cellular processes include cell survival, growth, differentiation, apoptosis and metabolism [10]. Previously, MKK7 has been demonstrated to play a cardio-protective role in the heart to mitigate heart failure [11-14]. However, it is unknown if it can also induce cardioprotection in the HFpEF heart.
In this study, we aim to ascertain whether therapeutically maintaining MKK7 expression holds cardioprotective benefits in terms of HFpEF. Using a two-hit mouse model of HFpEF, we first demonstrated the downregulation of both MKK7 and its activation in the myocardium. Therapeutically maintaining MKK7 expression, via adeno-associated virus (AAV) 9, mitigated cardiac diastolic dysfunction and reduced ventricular compliance. In terms of the underlying mechanism, it was observed that upregulation of cardiomyocyte-specific MKK7 hindered ER stress by maintaining the expression of IRE1-XBP1, which would have otherwise been downregulated. Reduced CHOP expression was also observed following MKK7 overexpression. To summarise, we provide proof-of-concept evidence of the cardio-protective nature of MKK7 to maintain the IRE1-XBP1 signalling pathway and alleviate HFpEF.
2. Methods
2.1. Study Approval
All laboratory mice used in this study were maintained in a pathogen-free facility at the University of Manchester. Animal studies were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and were approved by the University of Manchester Ethics Committee.
2.2. Heart Failure with Preserved Ejection Fraction (HFpEF) Mouse Model
The mouse model described by Schiatterella et al.[6] was used to generate the HFpEF mice model. Briefly, 6 to 8-weeks-old male C57BL/6N mice were randomly allocated and maintained with unrestricted access to either standard chow (SDS, 801960) or 60% high-fat-diet (HFD, 824053, BioTech). N ω-nitro-l-arginine methyl ester (L-NAME, Sigma Aldrich) was supplied in the drinking water at a concentration of 0.6 g/L, after the pH was adjusted to 7.4. Water was changed every two days to ensure drug stability and efficiency. Each mouse was considered a biological replicate. No exclusions were made. Mice were euthanised by cervical dislocation while sedated.
2.3. Echocardiography
Cardiac function evaluation was performed using echocardiology. Mice were anaesthetised with 1.5% isoflurane mixed with 100% oxygen at 1.5 L/min rate. The Acuson Sequoia C256 system (Siemens) and Vevo 770 system (Visualsonics) were used to obtain transthoracic M-Mode and pulse wave Doppler ultrasound images. Parameters such as fractional shortening (FS%), left ventricle chamber and wall dimensions, diastolic function (IVRT, E/A) parameters, ejection fraction (EF%), left ventricular (LV) mass and relative wall thickness were measured or calculated.
2.4. Conscious Blood Pressure Measurements
For blood pressure evaluation, CODA volume pressure recording system (CODA, Kent Scientific) was used to take non-invasive systolic and diastolic blood pressure as well as mean blood pressure. Measurements were performed at the same time of day, and ten acclimation cycles were discarded before starting to record. At least ten measurements were averaged per mouse.
2.5. Glucose Tolerance Test
Tail blood was acquired for glucose tolerance test. Mice were fasted for 6 h, followed by intraperitoneal glucose injection (2 g/kg body weight). Blood glucose levels were measured at 30-min intervals over 2 h using Accu-Chek Aviva (Roche) glucometer.
2.6. Construction of adeno-associated virus-MKK7 (AAV9-TnT-MKK7)
The pSSV9-TnT-eGFP was modified by replacing GFP with Flag-tagged human MKK7 cDNA (DU48691, MRCPPUreagents). To obtain the recombinant AAV9-MKK7 virus and the control AAV9-eGFP virus, HEK293T cells were co-transfected with an adenoviral helper plasmid (pDG∆VP), pAAV2-9 Rep-Cap plasmid (p5E18-VD2/9) and the adeno-associated virus genome plasmids pSSV9-TnT-MKK7 and pSSV9-TnT-eGFP, respectively. Recombinant AAV9-MKK7 was purified by discontinuous iodixanol gradient and titrated by quantitative polymerase chain reaction (qPCR) relative to a standard curve. To overexpress MKK7, 1 × 1011 genomic particles were administrated by intravenous injection. AAV9-GFP was injected as the control.
2.7. Histology
Isolated hearts were first washed in PBS before being immediately fixed in 4% paraformaldehyde for 5 h at 4 °C. Next, they were washed in PBS for 30 min before undergoing dehydration in 50%, 70%, 90% and 100% ethanol for 2 h each. The hearts were then cleared in xylene overnight and embedded in paraffin. The paraffin blocks were sectioned into 5 μm thickness using Microtome (Leica). Before histological staining was performed, sections were dewaxed and rehydrated.
2.8. Haematoxylin and Eosin Staining (H&E)
The assessment of the cardiomyocyte cross-sectional area was performed by H&E staining. All tissue sections underwent deparaffinisation before being sequentially rehydrated in 100%, 70% and 50% IMS for 10 min each. Next, sections were incubated in Harris Haematoxylin (LAMB/230, RA Lamb Dry Chemical Stains) for 5 min and differentiated with acid alcohol for 10 s. The nuclei were counterstained using Eosin (6766007, Thermo Scientific) for 1 minute. After rinsing twice in ddH2O, sections were dehydrated then cleared in xylene for 20 min. Sections were mounted using Eukitt Quick-hardening mounting media (03989, Sigma). After the slides were imaged using 3D Histech Panoramic 250 Slide-scanner, the cardiomyocyte cross-sectional area for individual cardiomyocytes was analysed using ImageJ. On average, the ventricular cross-sectional area of 100 cardiomyocytes for each heart was analysed.
2.9. Masson’s Trichrome Staining
Masson’s trichrome staining was performed to assess the extent of cardiac fibrosis. Following dewaxing and rehydration as previously mentioned, tissue sections were treated with Bouin’s fixative (HT10132, Sigma) for 2 h, before being stained with Harris Haematoxylin for 5 min. After the sections were differentiated in acid alcohol solution for 10 s they were treated with red solution for 5 min (HT151, Sigma), then incubated with 2.5% phosphomolybdic acid (HT153, Sigma) for 15 min. Subsequently, the tissue sections were briefly rinsed in ddH2O before being treated with Aniline Blue Solution (B8563, Sigma) for 5 min to stain collagen fibres. Next, sections were incubated with 1% Acetic Acid for 1 min before being sequentially dehydrated, cleared and mounted with Eukitt as previously mentioned. The slides were imaged using 3D Histech Panoramic 250 Slide-scanner, the colour threshold function on ImageJ was used to calculate the areas of fibrosis from 10 fields per sample.
2.10. Electron Microscopy
To analyse changes in endoplasmic reticulum structure, transmission electron microscopy was performed. Freshly isolated heart samples were fixed overnight in 0.1 M HEPES buffer (pH 7.2) containing 2.5% glutaraldehyde and 4% paraformaldehyde. Next, tissues were processed in 0.1 M Cacodylate Buffer (pH 7.2) with 1.5% potassium ferrocyanide and 0.1% osmium tetroxide for 1 h, before being incubated with 0.1 M Cacodylate Buffer (pH 7.2) with 1% tannic acid for 1 h and finally 1% uranyl acetate or a further hour. Next, samples were dehydrated in ethanol and embedded in TAAB 812 resin before being polymerised at 60 °C for 24 h. Finally, the sections were cut with a Reichert Ultracut Ultramicrotome and examined using the Talos L120C transmission electron microscope at 100kV accelerating voltage. Images were taken with Gatan Orius SC1000 CCD camera.
2.11. Immunoblotting Analysis
Total protein was obtained using Triton lysis buffer (137 mmol/L NaCl, 20 mmol/L Tris, 0.1% w/v SDS, 2 mmol/L EDTA, 10% v/v glycerol, 1% Triton-X, 25 mmol/L glycerophosphate, 1 mmol/L Na3VO4, 1 mmol/L, 10 mM NAM, 1 μM TSA, 1x protein cocktail inhibitor, pH 7.4). The lysates were cleared by centrifugation at 14,000× g for 20 min. Protein concentration was determined using the Bio-Rad protein assay. 30 µg protein extracts were used for Immunoblot analysis with antibodies against MKK7 (4172, Cell Signalling), phospho-MKK7 (4171, Cell Signalling), Flag (MA1-91878, Thermofisher), JNK (9252, Cell Signalling), p38 (9212, Cell Signalling), ERK1/2 (9102, Cell Signalling), MKK4 (9152, Cell Signalling), GRP78 (ab21685, Abcam), IRE1 (ab37073, Abcam), phospho-IRE (ab48187, Abcam), ATF6 (ab37149, Abcam), PERK (3192, Cell Signalling), phospho-PERK (3179, Cell Signalling), XBP1s (24868-1-AP, Proteintech), eIF2a (9722, Cell Signalling), phospho-eIF2a (9721, Cell Signalling), ATF4 (10835-1-AP, Proteintech), CHOP (2895, Cell Signalling) and G-Beta (166123. Santa Cruz Biotechnology). Secondary antibodies anti-rabbit (7074, Cell Signalling) or anti-mouse (7076, Cell Signalling) HRP conjugates were used. Immune-complexes were detected using Amersham ECL Prime/Select detection reagents (RPN2232/RPN2235, Amersham) and the ChemiDoc MP System (BioRad).
2.12. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Trizol (Invitrogen) was used to extract total RNA from cells and ventricular tissues. Next, samples were treated with DNAse (DNA-free Removal Kit, Invitrogen) to eliminate DNA contamination. RNA was converted into cDNA using Lunascript (NEB3010, New England Biolabs). All primers were purchased from Qiagen and qPCRs were conducted using SYBR Select PCR Master Mix (4472908, Applied Biosystems) following the manufacturer’s instructions. qPCR reactions were run in the Step One Plus PCR System (Applied Biosystems), and fold change was calculated by the comparative Ct (ΔΔCt) method. mRNA levels were normalised to 18S expression.
2.13. Data Analysis
Data are presented as mean ± SEM and analysed using one-way or two-way ANOVA followed by post hoc tests where appropriate. Comparisons between two groups were performed using Student’s t test. For data with skewed distribution, non-parametric tests were used. Statistical analysis was performed using the GraphPad Prism 9 software and p values < 0.05 were considered statistically significant.
3. Results
3.1. MKK7 Reduction in the HFpEF Myocardium
In order to investigate the role of MKK7 during cardiac stress, we first examined the expression of MKK7 in a murine model of HFpEF. We used a recently developed preclinical model of HFpEF in which male mice are subjected to a two-hit model of hypertensive and metabolic stress elicited by N ω-nitro-L-arginine methyl ester (L-NAME), to inhibit constitutive nitric oxide synthases, coupled with high-fat diet for 15-weeks ( Figure 1A). This model successfully recapitulated the myriad features of human HFpEF. Upon examination of the myocardium, it was revealed that 15-weeks HFD+L-NAME led to a depletion in both total expression and phosphorylation of MKK7 in the myocardium compared to CHOW-fed control mice (Figure 1B and 1C). Therefore, an involvement of MKK7 in HFpEF progression is plausible.
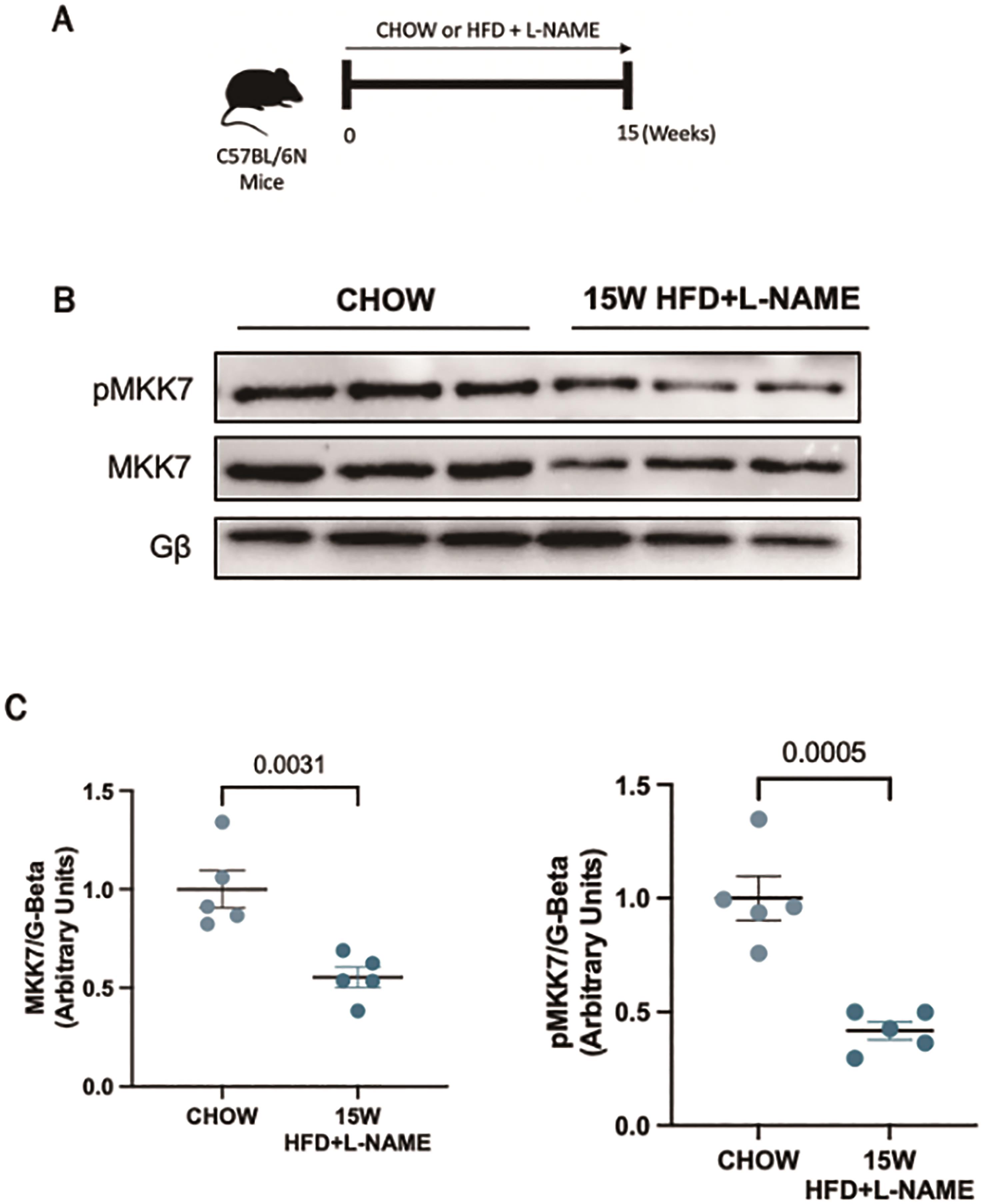
Figure 1. Downregulated MKK7 expression following 15-weeks HFpEF. (A) Experimental overview of HFpEF regime on male C57BL/6N mice for 15-weeks. (B + C) Immunoblotting analysis of total and phosphorylated MKK7 in male C57BL/6N mice myocardium following 15-weeks HFpEF (N=3)..
3.2. MKK7 Overexpression Mitigates Cardiac Dysfunction
To examine the role of MKK7 overexpression in HFpEF, mice were injected with AAV9-TnT-MKK7 before initiation of the HFpEF regime (Figure 2A and 2B). Before commencing with the study, the AAV9-TnT-MKK7 mouse model was characterised to ensure cardiac-specific MKK7 overexpression (Supplementary Figure S1A) As expected, 15-weeks HFD feeding led to increased body weight and glucose intolerance (Supplementary Figure S2A, S2B and S2C), while L-NAME exposure raised systolic and diastolic blood pressure (Supplementary Figure S2D and S2E). The level of systemic dysfunction between AAV9-TnT-GFP and AAV9-TnT-MKK7 HFpEF mice was comparable, implying both groups of mice experienced a similar degree of baseline metabolic dysfunction.
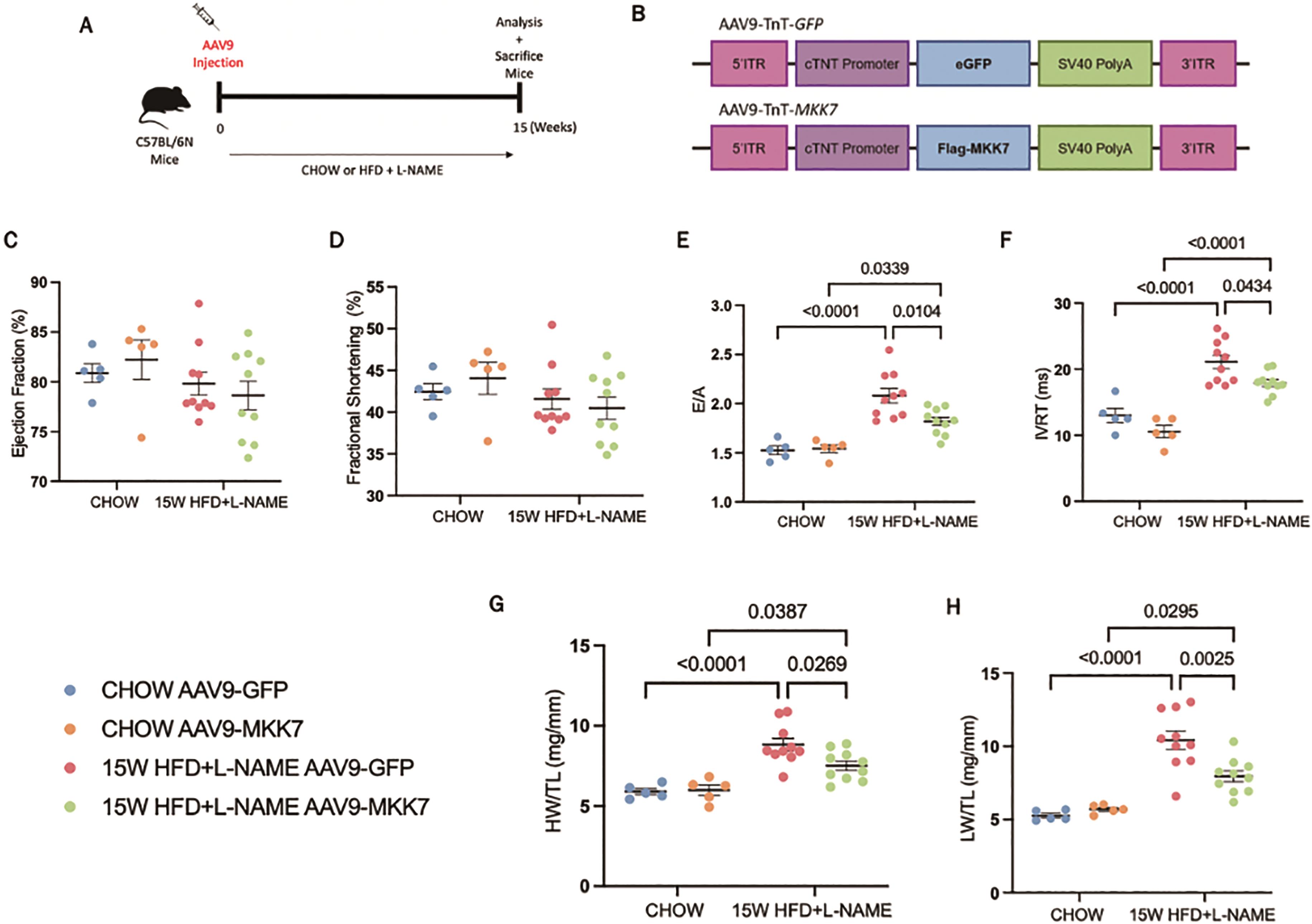
Figure 2. Cardiomyocyte-specific overexpression of MKK7 improved HFpEF phenotype. (A) Experimental overview of 15-weeks HFpEF on AAV9-TnT-eGFP or AAV9-TnT-MKK7 injected male C57BL/6N mice. (B) Construction of AAV9-TnT-eGFP and AAV9-TnT-MKK7 virus (C, D, E, F) Fractional shortening (FS%), Ejection fraction (EF%), E/A ratio, and IVRT of different experimental cohorts. (N=5‒10). (G) Heart weight/tibia length (N=5‒10). (H) Lung weight/tibia length (N=5‒10). Data presented as means ± SEM..
Echocardiography analysis was performed to evaluate changes in cardiac function. It was revealed that both AAV9-TnT-GFP and AAV9-TnT-MKK7 HFpEF mice had persistent preservation of ejection fraction and fractional shortening at the 15-week time-point (Figure 2C and 2D). However, markers of diastolic dysfunction such as IVRT and E/A ratio were significantly increased in AAV9-TnT-GFP HFpEF mice, but this response was mitigated in MKK7 overexpression mice evident by a blunted rise in E/A ratio and reduced IVRT length (Figure 2E and 2F). Furthermore, heart weight/tibia length (HW/TL) and lung weight/tibia length were significantly increased in AAV9-TnT-GFP HFpEF mice suggesting cardiac hypertrophy and pulmonary congestion (Figure 2G and 2H). This response was less prominent with MKK7 overexpression. At a cellular level, H&E staining analysis revealed that MKK7 overexpression alleviated pathological hypertrophic growth demonstrated by a smaller growth in cardiomyocyte cross-sectional area following HFpEF (Figure 3A and 3B). In a consistent finding, the mRNA expression levels of hypertrophic gene ANP, BNP and Myh7 were shown to be significantly lower in AAV9-TnT-MKK7 HFpEF mice compared to AAV9-TnT-GFP HFpEF mice (Figure 3C). Concomitantly, pathological interstitial and perivascular myocardial fibrosis was prevented by MKK7 overexpression, despite the HFpEF regime (Figure 3D and 3F). We also observed a slight increase in cardiomyocyte apoptosis in AAV9-TnT-GFP HFpEF mice but less so in AAV9-TnT-MKK7 (Figure 3G and 3H). Collectively, these results support the notion that MKK7 protects the heart against cardiac dysfunction.
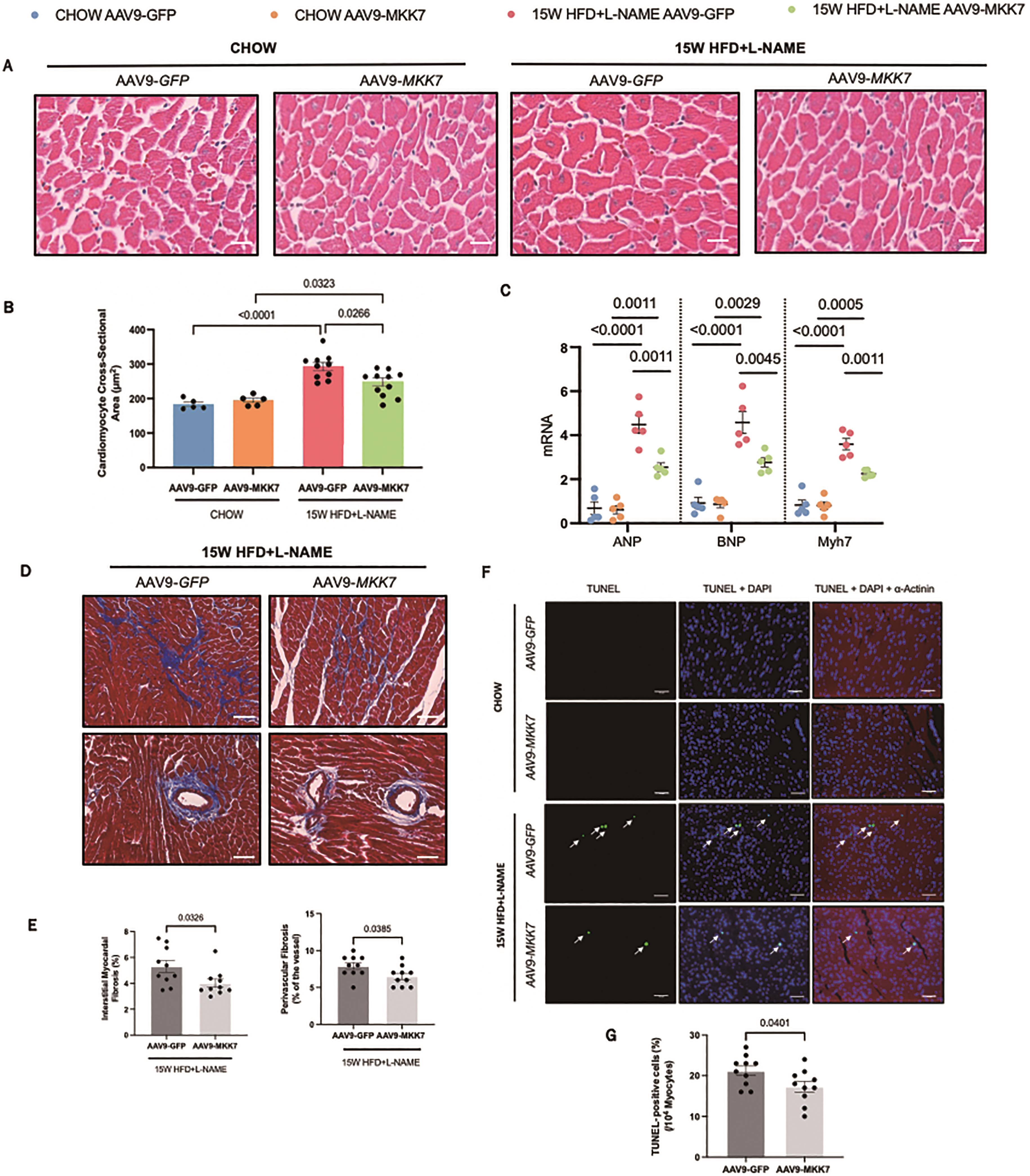
Figure 3. Overexpression of MKK7 prevented pathological ventricular remodelling, interstitial fibrosis and cardiomyocyte apoptosis during HFpEF stress. (A and B) H&E staining analysis of the cardiomyocyte cross-sectional area from different groups. Scale bar: 50 µM. (N=5‒10). (C) Relative mRNA expression of ANP, BNP and Myh7 (N=5). (D) Masson’s Trichrome staining of interstitial and perivascular fibrosis. Scale bar: 50 µM. (N=5‒10). (E) DHE staining. Scale bar: 50 µM. (N=5‒10). (F) Masson’s Trichrome staining analysis of interstitial and perivascular fibrosis. (G and H) TUNEL staining of the myocardium from different groups. Scale bar: 50 µM. (N=5‒10). Data presented as means ± SEM..
3.3. MKK7 Overexpression Alleviates ER Stress
Next, we attempted to gain clues regarding how MKK7 confers cardio protection. Accumulation of misfolded/unfolded proteins has recently been shown to occur in murine and clinical HFpEF, therefore to further explore this notion we evaluated changes in the unfolded protein response (UPR). This evolutionarily conserved adaptive response is activated upon ER stress to mitigate the disruption in protein quality control and preserve vital cardiac function. Accordingly, we assessed key UPR markers to appraise whether MKK7 regulates the UPR in the myocardium during HFpEF. It was noted that cardiac ATF6 and PERK-eIF2α-ATF4 signalling remained enriched at 15-week HFD and L-NAME in AAV9-TnT-GFP mice. In contrast, the expression of phosphorylated-IRE1, XBP1 splicing and GRP78 decreased following 15-weeks HFpEF, although total IRE1 expression remained constant. In addition, CHOP expression increased in the mouse myocardium after HFD and L-NAME treatment at 15 weeks (Figure 4A and 4B). Surprisingly, however, we detected preserved IRE1-XBP1s signalling together with reduced CHOP expression in AAV9-TnT-MKK7 HFpEF mice. Analysis of transcription levels revealed a general profile with decreased levels of GRP78, GRP94, and members of the ERAD pathway including HRD1, EDEM1, DERL3 and UFD1, in AAV9-TnT-GFP mice in response to 15-weeks HFpEF (Figure 4C). Analysis of the structure, using transmission electron microscopy, revealed slightly swollen ER AAV9-TnT-GFP HFpEF mice in comparison to AAV9-TnT-MKK7 HFpEF mice (Figure 4D). Together, these data demonstrate the ability of MKK7 in preserving IRE1-XBP1 signalling and reducing CHOP expression, suggesting a potential role for MKK7 in mitigating ER stress and maintaining protein quality control during HFpEF.
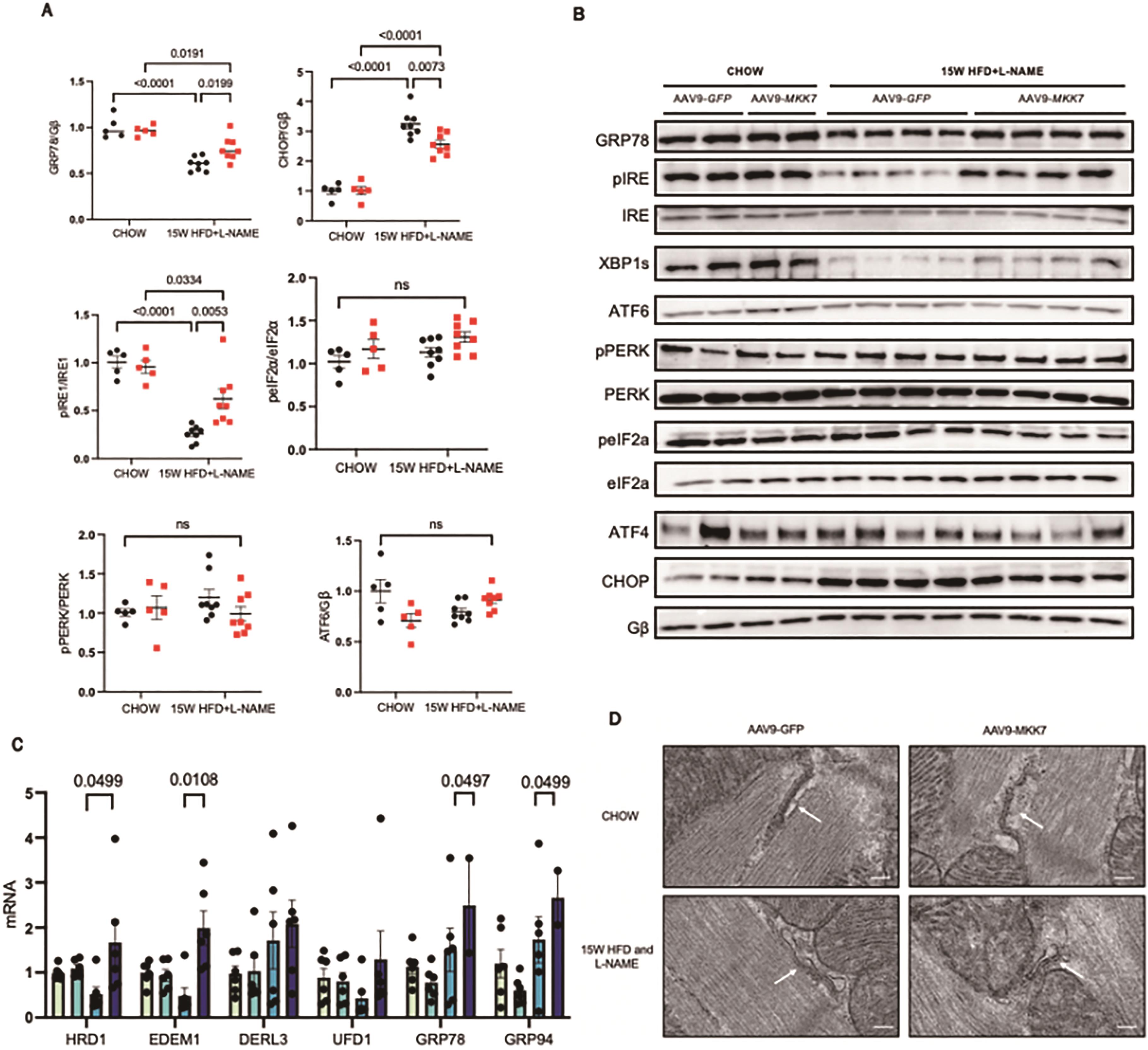
Figure 4. Mitigated ER stress and preserved IRE1-XBP1 signalling in MKK7 overexpression myocardium. (A and B) Representative immunoblot images and quantification of UPR markers in CHOW and 15-weeks HFpEF myocardium (N=5‒8). (C) Relative mRNA expression of components of the ERAD and UPR pathway in heart lysates from CHOW and HFpEF AAV9-TnT-eGFP and TAC or AAV9-TnT-MKK7 mice (N=5). Data presented as means ± SEM. (N=5). (D) Transmission electron microscopic images of the myocardium subjected to 15-weeks HFpEF in AAV9-TnT-eGFP or AAV9-TnT-MKK7 mice. Scale bar = 500 nm..
4. Discussion
In this study, we employed the use of a two-hit clinically relevant murine mouse model of cardiometabolic HFpEF. The key findings of this study are (1) downregulation of MKK7 in the HFpEF myocardium, (2) overexpression of MKK7 in the cardiomyocytes mitigates cardiac remodelling and diastolic dysfunction (3) mechanistically, MKK7 preserved IRE1-XBP1 signalling to mitigate ER stress. Our study provides novel insight into the clinical implications of using MKK7 in treating HFpEF.
The existing literature surrounding MKK7 in the context of the heart is inconclusive, with some studies suggesting it plays a detrimental function whereas others have demonstrated a cardioprotective effect. Although there have been some studies investigating the role of MKK7 during HFrEF [11-13] there have been no concise studies examining the role of MKK7 during HFpEF. Therefore, to investigate the link between MKK7 and HFpEF we used an experimental model of 60% high-fat-diet and L-NAME which has previously been shown to mimic the HFpEF phenotype [6,15]. The level of systemic dysfunction between AAV9-TnT-GFP and AAV9-TnT-MKK7 mice was comparable implying the mice experienced a similar degree of baseline metabolic dysfunction. This study highlighted that sustained HFpEF stress for 15-weeks dampened both MKK7 expression and activation. MKK7 is a stress-activated kinase that is rapidly activated following a stress insult. Reduced MKK7 expression following other forms of cardiac stress has also been identified such as long-term pressure-overload stress [11] and isoprenaline treatment [12]. This suggests that the lack of MKK7 in the heart following long-term stress has the potential to predispose mice to a range of pathologies. By maintaining MKK7 expression, via AAV9, this study hypothesised that it could protect the heart. Consequently, MKK7 overexpression was induced at the beginning of the study. A more therapeutic approach would be to induce MKK7 overexpression following the onset of the HFpEF, as this would be of more clinical relevance.
HFpEF is a disease lacking effective treatment options, primarily because of the lack of understanding regarding the mechanistic underpinnings of the disease. The endoplasmic reticulum is responsible for maintaining protein quality control by regulating protein synthesis and folding. ER stress occurs when there is an accumulation of misfolded proteins within the lumen of the ER due to cellular stress, such as oxidative stress [16] and hyperglycaemia [17]. To counteract the rise in misfolded protein the unfolded protein response (UPR) is initiated as an adaptive response. However, in the case of prolonged or severe stress, a pro-apoptotic phase is activated promoting cardiac dysfunction. Here, we unveiled that cardiomyocyte-restricted overexpression of MKK7 conferred cardioprotective properties by reducing fibrosis, cardiac hypertrophy and diastolic dysfunction. Although it is unknown how exactly MKK7 induced cardioprotection, we found that it is, at least partly, via preservation of IRE1-XBP1 signalling. No changes were observed in the PERK-ATF4 or ATF6 pathway, but analysis at earlier time-points such as 5-weeks would potentially identify any short-term fluctuations in the UPR signalling. In line with our study, it has been proven that the IRE1-XBP1 signalling pathway is downregulated during HFpEF. Schiatterella et al.[6] recently employed a similar two-hit mouse model to recapitulate the multi-organ HFpEF syndrome. They subjected mice to 60% high-fat-diet in combination with endothelial-dysfunction mediated hypertension and found a decline in myocardial IRE1 phosphorylation and XBP1s expression, accompanied by a decrease in diastolic function but stable systolic function. In a further study, the authors found that cardiomyocyte-specific overexpression of XBP1 rescued this pathological phenotype by reducing myocardial lipid accumulation It was found that mechanistically [15], XBP1 induced proteasomal-medial degradation of FoxO1 (Foxhead Box Protein 1) via the E3 ubiquitin ligase STUB1 (STIP1 homology and U-box-containing protein 1). However, changes in lipid accumulation were not examined in this study, therefore further research is warranted.
C/EBP homologous protein (CHOP) is a transcription factor involved in the UPR. Although the role of CHOP in other forms of HF has been extensively investigated, its role in HFpEF is not well known [18,19]. CHOP has been widely linked to the underlying processes involved in HFpEF, such as inflammation and fibrosis. Increased CHOP expression has been found to be present in obese mice resulting in increased levels of inflammatory cytokines promoting insulin resistance. Conversely, the depletion of CHOP prevented inflammation and insulin resistance [20]. CHOP expression has been widely associated with ER stress-induced cell death during metabolic stress [21,22] observed increased CHOP expression in a model of diabetic rats established by high-fat-diet feeding. However, these studies were not conducted in the setting of HFpEF. In this study, the expression of CHOP was significantly increased in AAV9-TnT-GFP mice fed HFD and L-NAME but less so in AAV9-TnT-MKK7 mice. This suggests that modulating CHOP expression may be a potential avenue to explore during the management of HFpEF.
Researchers are now identifying novel treatment options that limit the development and progression of HFpEF. Recently, attention has turn to sodium-glucose co-transporter 2 inhibitors, which have shown to reduce hospitalisation rates, and improve survival in numerous clinical trials investigating HFpEF. For instance, the sodium-glucose co-transporter 2 inhibitor, Empaglifozin, reduced the risk of cardiovascular disease related death and hospitalisation in HFpEF patients, regardless of the presence of diabetes [23]. Numerous other sodium-glucose co-transporter 2 inhibitors have demonstrated cardioprotective properties, including, Dapagliflozin [24] and Canaglifozin [25].
5. Limitations
A characteristic feature of HFpEF is that is occurs predominantly in obese woman, therefore, further research using female mice is warranted. Recently published data found that the use of this mouse model resulted in a phenotype that was more severe in male mice compared to female mice. In a study by Tong et al., the authors subjected both male and female mice to the HFD and L-NAME regime. They found that compared to male mice, female mice developed significantly less cardiac dysfunction demonstrated by normal heart mass and minimal changes in the E/e’ ratio. These changes were not mediated by female hormones as similar finding were found following ovariectomy [26]. Ageing is a defining characteristic of HFpEF, and studies have shown that older, obese woman are particularly susceptible to HFpEF. This increased risk can be attributed because with ageing the heart is more susceptible to remodelling with changes in structural and molecular processes that promote the onset of systolic and diastolic dysfunction [27]. Further research is required to investigate if MKK7 can still invoke a cardio-protective response in aged HFpEF mice.
Furthermore, left ventricular invasive haemodynamic data was not obtained in this study therefore, differences in intracardiac pressure level could not be determined. Also, this study unveiled that using a two-hit mouse model of HFpEF, there is downregulation of IRE1-XBP1 signalling cascade in the myocardium. This was found to be, at least partly preserved via cardiomyocyte-restricted overexpression of MKK7, however, the exact mechanism by which this was achieved is unknown.
6. Conclusion
Using a two-hit mouse model of HFpEF induced via 60% high-fat-diet in conjunction with nitric oxide synthase inhibitor L-NAME, this study examined the role of MKK7 in the development of HFpEF. Following 15-weeks of the HFpEF regime, we observed down regulation of MKK7 expression and activation. Meanwhile, overexpression of MKK7 maintain ER homeostasis to sustain protein quality control by preserving IRE1-XBP1 signalling. This mitigate pathological cardiac remodelling such as cardiac remodelling and fibrosis. This significantly improved diastolic dysfunction. Further research is warranted to identify methods to sustain MKK7 expression in the myocardium, leading to the formation of innovative approaches to tackle the development and progression of HFpEF.
Supplementary Materials: The following supporting information can be downloaded at: https://www. sciltp. com/ journals/ijddp/2024/1/337/216, Figure S1: Immunoblot analysis of cardiac-specific overexpression of MKK7 in C57BL/6N mice injected with AAV9-TnT-MKK7. (A) Validation of cardiomyocyte specific MKK7 overexpression in the myocardium of C57BL/6N mice injected with AAV9-TnT-MKK7 (N=1‒4); (B) Comparable levels of MKK7 expression in other tissues; Figure S2: 15-weeks of HFD and L-NAME feeding recapitulates features of clinical HFpEF. (A) Measurement of weekly body weight. (N=5‒10); (B and C) Measurement of glucose tolerance test and area under the curve (N=5‒10); (D and E) Data depicting systolic and diastolic blood pressure using non-invasive blood pressure. (N=5‒10) Presented as mean ± SEM.; Table S1: Structural parameters of the different mouse groups. End-diastolic interventricular septum thickness (dIVS), end-systolic interventricular septum thickness (sIVS), end-diastolic left ventricular internal diameter (dLVID), end-systolic left ventricular internal diameter (sLVID), end-diastolic left ventricular posterior wall thickness (dPW), end-systolic left ventricular posterior wall thickness (sPW), Left ventricular mass (LV mass). Data presented as mean ± SEM.
Author Contributions: T.A designed and carried out experiments, analysed and interpreted data, and wrote the manuscript. H.Z. acquired various data. S.H. & O.M. produced AAV9-TnT-MKK7. E.J.C. Designed animal works and reviews the study. X.W. conceptualized the project, designed experiments and interpreted results.
Funding: This study was supported by the British Heart Foundation (FS/19/39/34447) and a professorship from the German Centre for Cardiovascular Research (81Z0700201 to O.J. Müller). The authors wish to thank Roger Meadows, Steven Marsden, Peter March, and Darren Thomson (Bioimaging facility, University of Manchester) for technical training on microscopes; in addition to Aleksandr Mironov FBMH EM Core Facility (RRID:SCR_021147) for assistance and the Wellcome Trust for equipment grant support to the EM Facility (University of Manchester).
Institutional Review Board Statement: The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the University of Manchester (P2A97F3D, 2021).
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Acknowledgments: This study was supported by the British Heart Foundation (FS/19/39/34447) and a professorship from the German Centre for Cardiovascular Research (81Z0700201 to O.J. Müller). The authors wish to thank Roger Meadows, Steven Marsden, Peter March, and Darren Thomson (Bioimaging facility, University of Manchester) for technical training on microscopes; in addition to Aleksandr Mironov FBMH EM Core Facility (RRID:SCR_021147) for assistance and the Wellcome Trust for equipment grant support to the EM Facility (University of Manchester). The Histology Facility equipment used in this study was purchased with grants from The University of Manchester Strategic Fund.
Conflicts of Interest: All authors have declared that no conflict of interest exists.
References
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368‒378. DOI: https://doi.org/10.1038/nrcardio.2016.25
- Harper, A.R.; Patel, H.C.; Lyon, A.R. Heart failure with preserved ejection fraction. Clin. Med. 2018, 18, s24‒s29. DOI: https://doi.org/10.7861/clinmedicine.18-2-s24
- Upadhya, B.; Haykowsky, M.J.; Kitzman, D.W. Therapy for heart failure with preserved ejection fraction: current status, unique challenges, and future directions. Heart Fail. Rev. 2018, 23, 609‒629. DOI: https://doi.org/10.1007/s10741-018-9714-z
- Kjeldsen, S.E.; von Lueder, T.G.; Smiseth, O.A.; et al. Medical Therapies for Heart Failure with Preserved Ejection Fraction. Hypertension 2020, 75, 23‒32. DOI: https://doi.org/10.1161/HYPERTENSIONAHA.119.14057
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart. J. 2015, 36, 2585‒2594. DOI: https://doi.org/10.1093/eurheartj/ehv338
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351‒356. DOI: https://doi.org/10.1038/s41586-019-1100-z
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89‒102. DOI: https://doi.org/10.1038/nrm3270
- Azam, T.; Zhang, H.; Zhou, F.; et al. Recent Advances on Drug Development and Emerging Therapeutic Agents Through Targeting Cellular Homeostasis for Ageing and Cardiovascular Disease. Front. Aging 2022, 3, 888190. DOI: https://doi.org/10.3389/fragi.2022.888190
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. DOI: https://doi.org/10.1101/cshperspect.a011254
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; et al. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619‒1633. DOI: https://doi.org/10.1016/j.bbamcr.2010.12.012
- Chowdhury, S.K.; Liu, W.; Zi, M.; et al. Stress-Activated Kinase Mitogen-Activated Kinase Kinase-7 Governs Epigenetics of Cardiac Repolarization for Arrhythmia Prevention. Circulation 2017, 135, 683‒699. DOI: https://doi.org/10.1161/CIRCULATIONAHA.116.022941
- Liu, W.; Zi, M.; Chi, H.; et al. Deprivation of MKK7 in cardiomyocytes provokes heart failure in mice when exposed to pressure overload. J. Mol. Cell. Cardiol. 2011, 50, 702‒711. DOI: https://doi.org/10.1016/j.yjmcc.2011.01.013
- Liang, Q.; Bueno, O.F.; Wilkins, B.J.; et al. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 2003, 22, 5079‒5089. DOI: https://doi.org/10.1093/emboj/cdg474
- Shao, Z.; Bhattacharya, K.; Hsich, E.; et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ. Res. 2006, 98, 111‒118. DOI: https://doi.org/10.1161/01.RES.0000197781.20524.b9
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684. DOI: https://doi.org/10.1038/s41467-021-21931-9
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. doi: 10.3390/ijms18040771. DOI: https://doi.org/10.3390/ijms18040771
- Liu, B.; Zhang, Z.; Hu, Y.; et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc. Natl. Acad. Sci. 2019, 116, 21732‒21738. DOI: https://doi.org/10.1073/pnas.1907288116
- Fu, H.Y.; Okada, K.; Liao, Y.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361‒369. DOI: https://doi.org/10.1161/CIRCULATIONAHA.109.917914
- Miyazaki, Y.; Kaikita, K.; Endo, M.; et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1124‒1132. DOI: https://doi.org/10.1161/ATVBAHA.111.224519
- Suzuki, T.; Gao, J.; Ishigaki, Y.; et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell Rep. 2017, 18, 2045‒2057. DOI: https://doi.org/10.1016/j.celrep.2017.01.076
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381‒389. DOI: https://doi.org/10.1038/sj.cdd.4401373
- Ji, Y.; Zhao, Z.; Cai, T.; et al. Liraglutide alleviates diabetic cardiomyopathy by blocking CHOP-triggered apoptosis via the inhibition of the IRE-α pathway. Mol. Med. Rep. 2014, 9, 1254‒1258. DOI: https://doi.org/10.3892/mmr.2014.1956
- Anker, S.D.; Butler, J.; Filippatos, G.; et al. EMPEROR-Preserved Trial Investigators. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451‒1461.
- McMurray, J.J.V.; DeMets, D.L.; Inzucchi, S.E.; et al. DAPA-HF Committees and Investigators. The Dapagliflozin and Prevention of Adverse-outcomes in Heart Failure (DAPA-HF) trial: baseline characteristics. Eur. J. Heart Fail. 2019, 21, 1402‒1411. DOI: https://doi.org/10.1002/ejhf.1548
- Neal, B.; Perkovic, V.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 2099. DOI: https://doi.org/10.1056/NEJMc1712572
- Tong, D.; Schiattarella, G.G.; Jiang, N.; et al. NAD+ Repletion Reverses Heart Failure with Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629‒1641. DOI: https://doi.org/10.1161/CIRCRESAHA.120.317046
- Loffredo, F.S.; Nikolova, A.P.; Pancoast, J.R.; et al. Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ. Res. 2014, 115, 97‒107. DOI: https://doi.org/10.1161/CIRCRESAHA.115.302929


